
Abstract
The unsaturated zone plays a major role in elemental fluxes in terrestrial ecosystems. A representative chemical analysis of soil pore water is required for the interpretation of soil chemical phenomena and particularly to assess Trace Elements (TEs) mobility. This requires an optimal sampling system to avoid modification of the extracted soil water chemistry and allow for an accurate estimation of solute fluxes. In this paper, the chemical composition of soil solutions sampled by Rhizon® samplers connected to a standard syringe was compared to two other types of suction probes (Rhizon® + vacuum tube and Rhizon® + diverted flow system). We investigated the effects of different vacuum application procedures on concentrations of spiked elements (Cr, As, Zn) mixed as powder into the first 20 cm of 100-cm columns and non-spiked elements (Ca, Na, Mg) concentrations in two types of columns (SiO2 sand and a mixture of kaolinite + SiO2 sand substrates). Rhizon® was installed at different depths. The metals concentrations showed that (i) in sand, peak concentrations cannot be correctly sampled, thus the flux cannot be estimated, and the errors can easily reach a factor 2; (ii) in sand + clay columns, peak concentrations were larger, indicating that they could be sampled but, due to sorption on clay, it was not possible to compare fluxes at different depths. The different samplers tested were not able to reflect the elemental flux to groundwater and, although the Rhizon® + syringe device was more accurate, the best solution remains to be the use of a lysimeter, whose bottom is kept continuously at a suction close to the one existing in the soil.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The capacity of a soil to store and release chemicals influences crop production farm profitability and the quality of groundwater (Gish and Kung 2007). The flux of water at the bottom of the soil profile is an important parameter in many drainage studies. Unfortunately, the deep percolation through the bottom of the soil profile is difficult to assess (Kowalik 2006). To estimate the elemental flux, it is necessary to measure both the water flow and all element concentrations.
The moving water in soil can be split into gravitational water and capillary water. The gravitational water is the free water moving through the soil by gravity and is found in the macropores of the soil. On the contrary, the capillary water is held in the micropores of the soil by cohesion forces stronger than gravity.
The proportion of the two types of water may have a significant influence on the chemistry of draining water (Heinrichs et al. 1996). Indeed, Reynolds et al. (2004) have shown in a field study that concentrations of trace elements (TEs) were higher in soil solutions collected by suction samplers from those collected by zero-tension samplers. The latter samples preferentially macropores or bypass water (recent rainfall, irrigation events, preferential flow). As a significant part of the flow to groundwater occurs in homogeneous soils as capillary water (Di Bonito 2005), zero-tension lysimeters alone are not adapted to assess element fluxes to groundwater (Zhu et al. 2002).
As ceramic cups have been shown to both adsorb and release elements and thus change the chemistry of the soil solution, the “Rhizon®” soil moisture sampler has been preferred as it allows sampling without significantly disturbing the structure of the soil and the chemistry of the soil solution (Beesley et al. 2010; Di Bonito 2005; Duquette 2010; Moreno-Jiménez et al. 2011). However “Rhizon®” soil moisture samplers give values for a single point or a very small volume of soil, and thus do not take into account the textural and structural heterogeneity across a soil profile and consequently at field scale or larger (Armstrong et al. 1999). Moreover, they require an estimate of the drainage volume in order to calculate fluxes.
The main goal of this work was to find a sampling technique that may be able to assess as accurately as possible fluxes of various elements through the soil as compared to the flux measured at the bottom of a soil profile. In order to identify the various parameters that may be responsible for elemental composition and water flux variations, column experiments were performed with either pure sand or a mixture of sand and kaolinite, spiked or not with various elements. The pore water was collected by different suction samplers and compared to the results of the outlet of the columns. In order to identify the effect of moisture on the elemental composition of the soil solution, static substrate-water experiments were performed.
Materials and methods
Substrates
The substrates were prepared with Fontainebleau sand and the same sand mixed with pure kaolinite. Fontainebleau sand (type “Ga 39”) is composed of 98 % silica, whose saturated hydraulic conductivity in a column is K = 2.6.10−5 m s−1 with a pH of 7 ± 0.5 (50 g L−1, H2O, 20 °C). The sand was washed with nitric acid, rinsed with ultra pure water and dried in an air oven at 60 °C for 24 h before starting the experiment. Kaolinite was standard, obtained from Merck and consisted of about 96 % kaolinite and 4 % quartz (analyses were conducted using an X-ray diffractometer D500 (Siemens)), with a pH of 6.5 ± 0.5 (50 g L−1, H2O, 20 °C).
The metals added were chosen because they are among the most mobile ones and can present a threat to groundwater. The following metallic and metalloid minerals were used: Cr2K2O7, ZnCO3, Cr2O3, and As2S2. For As and Zn, the chemical forms were selected to reach solubility (standard solubility at pH 7) of a few millimolars, in order to provide continuous release. For Cr, this type of solubility was not found, so both soluble (Cr2K2O7) and fairly insoluble (Cr2O3) Cr minerals were chosen in order to provide gradual release. The addition of metallic minerals was made by weight of compound.
Column setup
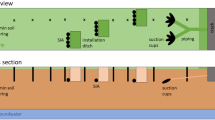
The column setup is shown in Fig. 1. PVC columns of 100 cm length and 7.6 cm inside diameter were closed at the bottom by a PVC cap with holes (diameter 6 mm). The polluted substrate (“topsoil”) was placed at the top 20 cm of the column (~2 kg, Fig. 1), the rest of the column was filled with the same material but non-spiked. Six columns were prepared, three with pure Fontainebleau sand (denoted as: S1, S2, S3) and three with 95 % of sand and 5 % kaolinite (denoted as: CS1, CS2, CS3). The top 20 cm of all columns (“topsoil”) was spiked with a mixture of Cr2K2O7, ZnCO3, Cr2O3, and As2S2 so as to reach element concentrations of 68, 80, 148, and 19 mmol kg−1, respectively, for Cr, Zn, Cr, and As.

Schematic view of the column experiment setup. left Classic column setup “S” and “P”; right Deviated flow setup “F”
In each column, 13 Rhizon® soil moisture samplers (5-cm-long Rhizon® samplers, Rhizosphere Research Products, the Netherlands) were installed at 15, 22, 25, 40, 60 (two Rhizon® per depth, with mixed solution) and 100 cm depth (three Rhizon® at 120° of each other). A depth of 0 cm corresponded to the surface of the “topsoil” layer. The samplers at 15, 22, and 25 cm were set to investigate changes in soil solution composition across the substrate layer boundary. Three Rhizon® samplers were installed at the bottom of the columns and connected to a vacuum vessel, to continuously collect the leachate by applying a constant suction (−100 mb). The three Rhizon® samplers were bulked together as one sample. Rhizon®s were acid-cleaned (0.01 M HNO3) prior to installation. The suction cups were inserted horizontally in the soil. Thus, the samples taken from these suction cups could be considered as an average soil solution at a given depth (soil volume sampled ca. 14 cm3 per Rhizon® (Duquette 2010)).
Vacuum types
Connected to the Rhizon® samplers, three types of suction were applied to each replicate group, that is: (i) syringe probes (“S”); (ii) vacuum samplers (“P”), and (iii) deviated flow (“F”).
Using 10-ml plastic syringes “S” applied to Rhizon® samplers, we measured with a vacuum gauge an average suction of −700 ± 10 mbar. The vacuum samplers “P” consisted of a centrifuge tube plugged with a rubber stopper sealed and perforated by two Tygon tubes (fixed pressure device). A tube maintained the vacuum while the second was connected to the Rhizon® probe. The applied vacuum was measured using a tensiometer and was close to −100 ± 5 mbar.
In order to sample the whole flow of solute through soil at each depth, we built a third setup called “deviated flow” “F”: the column was divided into five pieces where water was transferred from one to the other pieces by Rhizon® and Tygon tubings connected to a peristaltic pump (Fig. 1) (fixed flux device). This flux, despite a pump regulation, was different from the one in the other columns.
Two columns (S1 and CS1) were built using syringes, two columns (S2 and CS2) were built with half syringe samplers and the other half vacuum samplers, and two other columns (S3 and CS3) with the deviated flow.
Column experiments—protocol
The columns were successively irrigated with several additions of 160 ml of tap water (0.75, 0.58, and 1.53 mM of Ca, Mg, and Na, respectively) during 60 days. Four successive watering periods were applied: (1) the soil columns were watered with 160 ml per 24 h (corresponding to a rainfall of about 40 mm) in 4 days (160 ml × 4 times in 4 days); (2) the columns were watered with 160 ml per 48 h in 8 days (160 ml × 4 times in 8 days); (3) with 160 ml per 96 h in 16 days (160 ml × 4 times in 16 days); (4) 160 ml per 192 h in 32 days (160 ml × 4 times in 32 days). To avoid any evapotranspiration, the columns were closed at the top with aluminum paper.
Soil solution samplers (Rhizon®) in the profile were under-pressurized immediately after watering and sampled as soon as 1 mL was collected while the flow leaving the column (1-m deep) was sampled continuously. In the profile, the filling time for samplers spanned from 1 min to 24 h, as a function of depth. The total sampled volume in the profile was 10 mL (distributed over all profile Rhizon®) which represented less than 7 % of outlet.
The elemental fluxes were obtained by multiplying the water discharge by the measured concentrations. The water discharge was measured daily at the outlet while it was estimated to be equal to the watering amount at 22 and 25 cm. Fluxes presented here were those obtained at 22 and 25 cm, other depths gave similar trend values.
Static experiments
In order to investigate the effect of moisture on the elemental composition of the soil solution, four types of treatment were performed with the same elements as listed above: (1) a highly polluted Fontainebleau sand, with Cr2K2O7, ZnCO3, Cr2O3, and As2S2 with element concentration of 68, 80, 148, and 19 mol kg−1 for Cr, Zn, Cr, and As, respectively; (2) a lightly polluted sand, with Cr2K2O7, ZnCO3, Cr2O3, and As2S2 with element concentration of 0.68, 0.80, 14.8, and 0.19 mmol kg−1 respectively; (3) a mixture of sand + clay (equivalent to the previously used one) lightly; and (4) highly polluted.
Substrates (100 g) were saturated with tap water (100 %) then stabilized for 8 days at 20 °C to equilibrate the system. The substrates were not shaked during the experiment. The soil solution was sampled with a syringe connected to a Rhizon® by 5 ml successive steps until 50 % soil saturation was reached, the value which was the maximum that could be sampled by syringe.
Analyses
The pH in the solutions was determined with micro electrode (±0.02 pH units). The spiked elements (Cr, As, Zn) and the non-spiked elements (Ca, Mg, Na, Ni) in solution were measured by ICP-OES (Thermo Fisher, iCAP 4000, detection limits: As 0.026 μmol L−1, Ca 1.025 μmol L−1, Cr 0.0115 μmol L−1, Mg 0.29 μmol L−1, Na 2.66 μmol L−1, Zn 0.0046 μmol L−1), with a standard error of less than 5 % between three replicates. The water used for analyses was purified with Milli-Q Water Purification system (18 MΩ, Millipore Corporation).
Results and discussion
Static experiment
“S” samplers, by construction, collect water at higher depression, than the “P” and “F” ones. The static substrate-water equilibrium experiments were performed to see if the differential depression applied to columns might be responsible for differences in soil solution composition.
The pH of the solution collected was similar throughout the tested range of saturation (pH 7.5–8). Arsenic concentration in solution was close to 10 mg L−1 when the soil was quasi-saturated (95 %) and remained stable until 60 % saturation (Fig. 2). The trends of the results are quite similar for all elements (Cr, Zn), for both concentrations in both substrates except for the first 10 mL collected in the sand columns.

As concentration versus soil saturation obtained in static experiment
Despite differences between sand and sand + clay columns, which could be linked to the role of the clay particles; there was no effect of water content on the solution chemistry. In all cases, the concentrations in solution were the same for different water contents down to 50 % saturation, which was the lower limit for sampling with syringes.
Several authors suggest that water composition may be linked to suction (Gaudet et al. 1977). The results obtained in the static experiment show that As, Cr, and Zn (spiked elements) concentrations of solutions collected by “S” are constant for a soil water saturation between 50 and 90 % indicating that in our case water saturation was not responsible for concentration variation and that syringe linked to Rhizon would give consistent results over a significant range of soil water potentials. No link between water suction and composition can be established below this saturation, as syringe has not sampled this water.
In the static experiment, the water was immobile, giving time for the soil solution composition to equilibrate between all pore sizes, while this would not be the case for moving water as shown by Gaudet et al. (1997).
Water flux in the experimental columns
As our objective was to assess the influence of the sampling method on element fluxes, we first had to calculate the water flux at the outlet. Figure 3 shows the cumulative water flux during the experiment for the different columns and the cumulative flux of watering. The cumulative water fluxes after day 20 at the outlet are consistent with the input water recharge, and indicate that the temporal delay is not more than 5 days (during which there was water storage within the column); this phenomenon was also observed by Kasteel et al. (2007). Despite all precaution taken to avoid evapotranspiration and leaks, it appeared that the slope of cumulative fluxes was smaller than the watering flux after day 20. This may be due to the small amount of water added, increasing the relative importance of losses.

Cumulative water flux calculated at the outlet of the different column devices
Syringe samplers
To determine the variability within a substrate column, we compared the solution collected by the “S” probe with the solution collected at the outlet in the same column. We worked in cumulative fluxes as temporal interval and output volumes vary over time.
Non-spiked elements
The non-spiked elements are not present in the mixture of metals added. Because columns had been equilibrated with water before use and the water input was large (160 ml by day), we assumed that equilibrium between solution and minerals may have been reached during the experiment and thus fluxes in the profile should have been constant. If the flux in the profile was constant, then the outlet flux should be similar to the entering one. The cumulative fluxes of Ca, Mg, and Na calculated within the columns and at the outlet of the columns are presented in Fig. 4.

Cumulative fluxes of Ca, Mg, and Na obtained by “S” sampler as a function of time compared to the cumulative flux of water recharge
In sand columns, Na did not seem to interact with the substrates, as the outlet flux was quasi-equal to the Na flux added with watering. For Mg, the fluxes at 22 and 25 cm were similar to the outlet but higher than the watering flux.
For Ca and Mg in sand + clay column, the outlet flux was much higher than the watering flux and fluxes at 22 and 25 cm. This was even more obvious for Na in sand + clay, where fluxes at 22 and 25 cm were significantly higher than watering and flux at the outlet. These higher fluxes shall be linked to a release of these elements by clay.
However, because most of the variation in elemental concentrations occurred before day 24, fluxes calculations were recalculated starting from day 24 to compare stabilized fluxes. Following this assumption, in the sand system, cumulative fluxes for the outlet and at 22 and 25 cm were similar for all the elements analyzed, while large differences were observed in the sand + clay system (Fig. 5).

Cumulative fluxes of Ca, Mg, and Na obtained by syringe from day 24 of the experiment, in sand column and in sand + clay columns
Spiked elements
For spiked elements, we could not define a priori the outlet flux. In addition, an equilibration time was necessary to assess flux. Therefore, we only made the comparisons of the fluxes after stabilizing phases starting from the day 24 (Fig. 6).

Cumulative fluxes of As, Cr, and Zn obtained by syringe from day 24 of the experiment, in sand and sand + clay columns
The results after equilibration time showed that:
-
All Cr fluxes in sand were similar showing the tracer behavior of anionic Cr and accurate sampling.
-
For As in sand + clay columns, fluxes at 22 and 25 cm were similar and much higher than outlet fluxes. This behavior might be explained by sorption along the column.
-
For Zn in both columns, there was a large difference between 22 and 25 cm fluxes, questioning the capacity of syringe samplers to get reliable Zn concentrations.
-
For As in sand, and to a minor extent for Cr in sand + clay column, the outlet flux was clearly higher than fluxes at 22 and 25 cm. Knowing that water flux at the outlet was similar to that of the watering (Fig. 3) and the 22 and 25 cm sampling (see section Materials and methods) and that the columns were not contaminated below 22 cm, this difference may have only resulted from sampling under-concentrated water at 22 and 25 cm depth.
Identification of maximal concentration
In order to assess the ability of the sampler to sample peak concentrations, we examined the maximum concentrations obtained regardless of the time of occurrence.
Chromium, As, and Zn maximum concentrations obtained by “S” in sand and sand + clay columns, are presented in Table 1. Peaks presented here corresponded to high concentrations during the first leaching volumes passed through the spiked layers.
Within sand columns, the maximum chromium concentration at the bottom of the spiked zone (22 or 25 cm) was equal to 34.2 mM, whereas it was 59.9 mM at the outlet (100 cm). Moreover, the peak at 40 cm (53.3 mM) was much higher than the one at 22 cm (1780 mg L−1). This difference was also observed for arsenic, which had a maximum (peak) concentration in the column of 0.043 mM while it was higher at the output (0.052 mM). For zinc, the effect is the opposite: peak concentrations in the columns (2.38 mM) were higher than at the outlet (1.26 mM).
The variation of concentration values at the peak and at different depths might be a consequence of this absence of equilibrium under flowing conditions.
In sand + clay columns, concentrations were significantly higher than in sand. The maximum Cr, Zn, and As concentrations were, respectively, 135.5, 2.99, and 0.141 mM in the profile whereas it was 15.9, 0.12, and 0.0.13 mM at the outlet of the column.
In the first case, we can assume that the clay substrate should adsorb trace elements (As, Cr, and Zn) as has often been observed (Takahashi et al (1999)). Because of this adsorption, the flow of As, Cr, and Zn should be lower in the clay substrate with respect to the stream obtained in sand substrate alone. But the results obtained in the column (Fig. 6) and the maximum concentration (Table 1) show that the concentrations obtained in the clay substrate are larger than that of the substrate obtained in sand.
We believe that due to the higher capacity of the water retention clay substrate, the contact time between the contaminated area and the solution is higher in the clay substrate relative to the substrate-only sand. This allows time for the reaction of dissolved minerals to occur, thus increasing the flow of As, Cr, and Zn in the profile.
Comparison of methods
Non-spiked elements
A significant number of samples from “P” or “F” devices were lacking water, leading to a difficulty to assess precisely the fluxes. Figure 7 presents the calculated cumulative fluxes from day 24 of the experiment for the two specific samplers, like for the “S” device; fluxes were stabilized after 24 days.

Cumulative flux of Ca, Mg, and Na obtained by “F” and “P” from day 24 of the experiment, in sand column and in sand + clay columns
We observed three different patterns:
-
i.
The outlet fluxes were similar to the 22 and 25 cm fluxes. This was the case for Mg and Na sampled with the “F” device.
-
ii.
The outlet fluxes were lower than the 22 and 25 cm fluxes. This is the case for Ca sampled by “F” device in sand and for Na sampled by “P” device in sand + clay column.
-
iii.
There was a high variability between all fluxes, for all elements sampled by the “P” device.
These values tend to show that “P” and “F” samplers, although presenting different approaches, do not perform better than the classical syringe, while “S” shows less variability (Fig. 5).
Spiked elements
The results of “F” and “P” samplers for spiked elements (Fig. 8) showed even more differences between sampling locations than non-spiked elements. Only Cr in sand + clay columns sampled by “P” sampler showed some consistency. Some fluxes could not be calculated due to lack of sampled solution. The syringe therefore remains the best way to sample.

Cumulative flux of As, Zn, and Cr obtained by “F” and “P” from day 24 of the experiment, in sand column and in sand + clay columns
Flux estimation
Cumulative fluxes obtained by syringe at various depths after a stabilization period were compared to the flux at the outlet of the column. It appears that in sand, and for non-spiked elements, the syringe samplers provided correct estimates of concentrations as the calculated flux were similar to the one measured at the outlet. It is worth mentioning that during the first 24 days the fluxes varied largely between columns and between input and output. A significant equilibration time is thus necessary before any flux estimation. This time corresponds to an equivalent of approximately 500 mm of infiltrated water. It also corresponds to the time at which the irrigation rate was significantly decreased. Therefore, the differences between input and output observed during the first phase could be linked to the effect of rapid flow.
For elements spiked as solids in the top 20 cm of the column, the flux estimation showed significant deviation for As and Zn concentrations. This can be partly explained by the peak values of concentrations. Indeed, due to rapid flow during a short time, despite syringes sampling just after the irrigation events, it appeared that the syringe could not sample peak values of concentration accurately. As shown by Bloem et al. (2009) in hydrophilic soil without cracks or other obvious causes of preferential flow, the spatial distribution of drainage and solute transport indicate markedly nonuniform flow in a soil. So, the difference observed sometime between 22 and 25 cm could be explained by a nonuniform flow in the column.
This phenomenon is explained by McGuire and Lowery (1994) who showed that the sampled water does not correspond exactly to the moving one and needs to be corrected by instantaneous local water flux to get representative fluxes. Under these conditions, the flux estimation gave the range of error to be higher than a factor of two in our experiments. This difference between porous cups sampling and outflow, although significant, appears much smaller than the one occurring in field conditions (Stutter et al. 2005).
In sand + clay columns, it is difficult to compare the flux estimates at 22–25 cm depth with the outflow because sorption/desorption phenomena could occur on the clay that is present below the spiked layer. Ion exchange has not been identified as all concentrations are increasing or decreasing. However, adsorption is particularly important for Mg and As, leading to variation of fluxes by a factor higher than 5. Due to this sorption and the setup of the experiment, it was not possible to determine if the sampler played any role on the solution chemistry in this type of medium.
All concentrations were significantly higher in sand + clay columns than in sand ones. This could be related to the fact that peak values cannot be accurately sampled in sand columns. Indeed, the water may flow more slowly in sand + clay columns and thus the sampled water might be closer to the moving one.
Conclusion
The main goal of this work was to find a sampling technique that may be able to assess as accurately as possible the fluxes of various elements through the soil as compared to the flux measured at the bottom of a soil profile. In order to identify the various parameters that may be responsible for elemental composition and water flux variations, column experiments were performed with either pure sand or a mixture of sand and kaolinite, spiked with chromium, arsenic, and zinc. The pore water was collected by different suction samplers and compared to the results of the outlet of the columns. Three methods for soil solution sampling connected to Rhizon® have been used and compared: the syringe (“S”) sampler, the vacuum sampler (“P”), and the deviated flow (“F”).
It appears that in sand, and for non-spiked elements, the syringe samplers provided correct estimates of concentrations as the calculated flux were similar to the one measured at the outlet.
For elements spiked, the flux estimation showed significant deviation for As and Zn concentrations. It appeared that the syringe could not sample peak values of concentration accurately. And water does not correspond exactly to the moving one and needs to be corrected by instantaneous local water flux to get representative fluxes. Under these conditions, the flux estimation gave range of error higher than a factor of two to five in our experiments.
The results show that samplers either operating at fixed pressure (“P”) or at fixed flux (“F”) do not provide better results than syringe (“S”). The syringe therefore remains the best way to sample.
From a general point of view, our results emphasize the need for the use of lysimeters, which has already been advised in numerous papers. However, if one accepts an error of a factor of two, the syringe sampler may provide estimates of element fluxes. Other samplers with specified suction or flux do not provide better results. This study also allowed a better understanding of the effect of water saturation on solution chemistry. When part of the water flows rapidly, it is fairly difficult to sample it and the sampled water is an undefined mixture between fast- and slow-flowing water.
References
Armstrong, A. C., Leeds Harrison, P. B., Harris, G. L., & Catt, J. A. (1999). Measurement of solute fluxes in macroporous soils: techniques, problems and precision. Soil Use and Management, 15(4), 240–246.
Beesley, L., Moreno-Jiménez, E., Clemente, R., Lepp, N., & Dickinson, N. (2010). Mobility of arsenic, cadmium and zinc in a multi-element contaminated soil profile assessed by in-situ soil pore water sampling, column leaching and sequential extraction. Environmental Pollution, 158(1), 155–160.
Bloem, E., Hogervorst, F. A. N., & de Rooij, G. H. (2009). A field experiment with variable-suction multi-compartment samplers to measure the spatio-temporal distribution of solute leaching in an agricultural soil. Journal of Contaminant Hydrology, 105(3–4), 131–145.
Di Bonito, M. (2005). Trace elements in soil pore water: a comparison of sampling methods. University of Nottingham.
Duquette, M.-C. (2010). Mesure de la concentration en métaux traces dans la solution de sol par la microlysimétrie. Université de Montréal: Faculté des études supérieures et postdoctorales.
Gaudet, J. P., Jégat, H., Vachaud, G., & Wierenga, P. J. (1977). Solute transfer, with exchange between mobile and stagnant water, through unsaturated sand. Soil Science Society of America Journal, 41(4), 665.
Gish, T. J., & Kung, K.-J. S. (2007). Procedure for quantifying a solute flux to a shallow perched water table. Geoderma, 138(1–2), 57–64.
Heinrichs, H., Böttcher, G., Brumsack, H.-J., & Pohlmann, M. (1996). Squeezed soil-pore solutes—a comparison to lysimeter samples and percolation experiments. Water, Air, & Soil Pollution, 89(1), 189–204.
Kasteel, R., Pütz, T., & Vereecken, H. (2007). An experimental and numerical study on flow and transport in a field soil using zero tension lysimeters and suction plates. European Journal of Soil Science, 58(3), 632–645. doi:10.1111/j.1365-2389.2006.00850.x.
Kowalik, P. J. (2006). Drainage and capillary rise components in water balance of alluvial soils. Agricultural Water Management, 86(1–2), 206–211.
McGuire, P. E., & Lowery, B. (1994). Monitoring drainage solution concentrations and flux in unsaturated soil with a porous cup sampler and soil moisture sensors. Ground water.
Moreno-Jiménez, E., Beesley, L., Lepp, N. W., Dickinson, N. M., Hartley, W., & Clemente, R. (2011). Field sampling of soil pore water to evaluate trace element mobility and associated environmental risk. Environmental Pollution, 159(10).
Reynolds, B., Stevens, P. A., Hughes, S., & Brittain, S. A. (2004). Comparison of field techniques for sampling soil solution in an upland peatland. Soil Use and Management, 20(4), 454–456.
Stutter, M. I., Deeks, L. K., & Billett, M. F. (2005). Transport of conservative and reactive tracers through a naturally structured upland podzol field lysimeter. Journal of Hydrology, 300(1–4), 1–19.
Takahashi, Y., Minai, Y., Ambe, S., Makide, Y., & Ambe, F. (1999). Comparison of adsorption behavior of multiple inorganic ions on kaolinite and silica in the presence of humic acid using the multitracer technique. Geochimica et Cosmochimica Acta, 63(6), 815–836.
Zhu, Y., Fox, R. H., & Toth, J. D. (2002). Leachate collection efficiency of zero-tension pan and passive capillary fiberglass wick lysimeters. Soil Science Society of America Journal, 66(1), 37–43.
Acknowledgments
This study was funded by Innovasol Foundation and ADEME.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Coutelot, F., Sappin-Didier, V., Keller, C. et al. Comparison of soil solution sampling techniques to assess metal fluxes from contaminated soil to groundwater. Environ Monit Assess 186, 8929–8941 (2014). https://doi.org/10.1007/s10661-014-4055-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10661-014-4055-4