
Abstract
The diversity of Phakopsora pachyrhizi populations has often resulted in the selection of virulent pathotypes following the release of rust-resistant cultivars. Thus, knowledge of the pathogenicity and variability of P. pachyrhizi specie, coupled with an understanding of its biology and host-pathogen interactions, is essential for better comprehension of the evolution and rapid adaption of the pathogen. Such comprehension can be useful for genetic breeding programs and other research areas. Therefore, this study aimed to assess the genetic variability and virulence among P. pachyrhizi mono-uredinial isolates obtained from field samples collected in Brazil during the period from 2007 to 2012 by comparing the sequences of Internal Transcribed Spacer (ITS) regions and AFLP markers and analyzing the responses of differential soybean genotypes. The polymorphisms among the AFLP markers (locally) and ITS sequences (globally) demonstrated the high genetic diversity of this pathogen. Phenotypic analysis revealed the presence of at least 16 different pathotypes present in soybean fields in Brazil. The most effective resistance gene was Rpp5 (PI 200487), followed by Rpp4 (PI 459025) and Rpp2 (230970). This study represents the first report on the virulence and molecular characterization of a collection of P. pachyrhizi mono-uredinial isolates, which might represent different races occurring in Brazilian fields in the period from 2007 to 2012.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Asian soybean rust is currently the most severe soybean (Glycine max) disease and is caused by Phakopsora pachyrhizi Sydow & Syd. This biotrophic basidiomycete threatens soybean production all over the globe, but the threat is most severe in the major soybean growing areas in South America (Langenbach et al. 2015). In Brazil, the disease was first observed in 2001, and since then, it has become one of the most serious economic threats given its potential to reduce yields (Yorinori et al. 2005).
P. pachyrhizi naturally infects 31 species in 17 legume genera. This broad range of hosts may contribute to a diverse and complex pattern of virulence presented by the pathogen (Frederick et al. 2002; Hartman et al. 2005).
In soybean, resistance genes to P. pachyrhizi, named Rpp1, Rpp2, Rpp3,
Rpp4, Rpp5, Rpp6 and Rpp7, are described in the literature (Bromfield and Hartwig 1980; Mclean and Byth 1980; Hartwig and Bromfield 1983; Hartwig 1986; Garcia et al. 2008; Li et al. 2012; Childs et al. 2018). However, these genes are considered race-specific and there are numerous reports of their ineffectiveness when genotypes are infected with fungal isolates from different continents or isolates that do not contain the corresponding virulence gene (Yamaoka et al. 2002, 2010; Milles et al., 2011; Akamatsu et al. 2013, 2017), indicating the existence of genetic and virulence variability in fungus populations that is reflected in the diversity of races.
Molecular tools have been effectively used in the study of the genetic variability of many organisms, including fungi. Because they evolve rapidly and thus exhibit high intraspecific variation in their nucleotide sequences and length, and because they can be easily amplified via PCR (Polymerase Chain Reaction), the transcribed internal spacer regions (ITS) have been widely used to obtain an indication of the variability present in a species or at the population level (Cruz et al. 2006; Martin 2007). AFLPs.
(Amplified fragment length polymorphisms) are a firmly established type of molecular marker for assessing intraspecific genetic diversity, considering their robustness and the high numbers of reproducible polymorphic bands that can be obtained with just a few primer and/or enzyme combinations (Vos et al. 1995; Meudt and Clarke 2007).
Asian soybean rust management requires knowledge about pathogen biology and host - pathogen interactions and studies on new sources of resistance to the pathogen. Although the genetic diversity presented by P. pachyrhizi populations has been identified over the years in several studies, showing the existence of an efficient mechanism of dispersion and gene flow as well as temporal and geographical pathogenic variation (Freire et al. 2008, 2012; Twizeyimana et al., 2011; Zhang et al. 2012; Akamatsu et al. 2013, 2017; Jorge et al. 2014), no previous studies have focused on genetic diversity and its correlation with virulence in P. pachyrhizi mono-uredinial isolates.
Therefore, the use of pure isolates is fundamental in investigations of the role of specific genes in the infection process and in host - pathogen interactions to ensure greater reliability in phenotypic evaluations, allowing the accurate determination of resistance and susceptibility reactions in the evaluated genotypes. The dikaryotic nature of the urediniospores of P. pachyrhizi promotes the maintenance of sequence variations and heterozygosity at different levels among the genome; each nucleus may preserve distinct repeat motifs within a single urediniospore. In this way, pure isolates can be helpful to understand the mechanisms involved in resistance and sensitivity to Rpp genes and fungicides, because mutations can be monitored at the individual level in terms of zygosity and expression levels between the two nuclei, allowing, for example, studies of fitness costs.
Thus, the aims of this study were to obtain and analyze the genetic diversity of P. pachyrhizi mono-uredinial isolates obtained from samples collected in different Brazilian soybean producing regions and in different years (2007–2012) by comparing the ITS sequences and AFLPs locally and globally (with sequences available in NCBI) and correlating these markers with virulence responses after infection of a soybean differential set containing different Rpp profiles, in order to evaluate how diverse is the collection of isolates obtained and potential races.
The knowledge of the virulence spectrum and variability of the pathogen are fundamental to better understanding the pathogen adaptability and the capacity of isolates to overcome resistance mediated by the Rpp genes. In addition, such knowledge provides a foundation to identify important genetic sources of resistance to be explored by breeding programs.
Materials and methods
Samples and pathogen isolates
The biotrophic nature of P. pachyrhizi hinders its in vitro manipulation. Therefore, the purification and characterization of P. pachyrhizi isolates are laborious tasks. The use of detached leaves is a routine method frequently used to overcome the limitations of time and space in greenhouses, especially when working with purified isolates (Twizeyimana and Hartman 2010). The use of this methodology enables the production and maintenance of mono-uredinial cultures in a practical and efficient way to avoid any risk of contamination between the different isolates.
Leaf samples infected with P. pachyrhizi that were collected in different soybean producing regions of Brazil (Fig. 1) were used to obtain the isolates by means of a mono-uredinial culture on detached leaves. Spores of a single uredinium were transferred to healthy leaves of the susceptible genotypes Embrapa 48 and Coodetec 219RR (CD219RR), which were kept in a Petri dish containing 1% agar-water medium, and then maintained in a growth chamber with a 14-h photoperiod, a temperature of approximately 21 °C and a humidity level of 60% for approximately 15 days for the production of new spores. To guarantee the purity of the isolates, three cycles of mono-uredinial culture were carried out on detached leaves. Detached leaves of the same susceptible genotypes were used for spore multiplication, and the spores were transferred to a 2 mL microtube kept open in a desiccator for 24 h for dehydration of the spores and were then stored in a freezer at −80 °C for spore preservation. Regarding P. pachyrhizi DNA extraction, fresh spores were used to study the genetic variability of the isolates, and frozen spores were used for phenotypic evaluation after inoculation in a group of soybean differentials.

The sampling locations of Asian soybean rust isolates collected in Brazil are shown on the map. States from which pathogen samples were obtained are indicated in dark gray
DNA extraction and PCR amplification
The total genomic DNA of each isolate was extracted from a suspension of spores kept in Petri dishes containing 1% autoclaved agar-water for approximately 16 h at 21 °C in the dark. With the aid of a glass Drigalski handle, the spores were collected and transferred to 1.5 mL microtubes and centrifuged for 5 min at 12,000 rpm. The supernatant was discarded, and the precipitate was used for DNA extraction following a protocol described by Dellaporta et al. (1983), with modifications. After extraction, the DNA was quantified in a NanoDrop® ND-1000 UVVis spectrophotometer, and its integrity was verified by means of 0.8% agarose gel electrophoresis. Finally, the samples were diluted to a final concentration of 10 μg/μL per sample. The DNA was stored at 4 °C.
For the specific detection of the P. pachyrhizi fungus, the extracted DNA was subjected to PCR with the oligonucleotides Ppa1 (5’-TTAGATCTTTGGGCAATGGT3’) and Ppa2 (5’-GCAACACTCAAAATCCAACAAT-3′), which amplified a 332 bp fragment corresponding to the ITS1–5.8S-ITS2 region of the genome of P. pachyrhizi (Frederick et al. 2002). For final PCR amplification, the reaction mixture comprised 50 ng of genomic DNA in a 25 μL reaction volume containing 1X buffer, 200 μM dNTPs, 1.5 mM MgCl2, 0.25 U of Taq DNA Polymerase (Invitrogen) and 100 pmol of each oligonucleotide. The PCR program was 5 min at 94 °C followed by 35 cycles of 60 s at 94 °C for template denaturation, 60 s at 55 °C for primer annealing, and 120 s at 72 °C for primer extension, plus a final extension step of 7 min at 72 °C. The product of the amplification reaction was then separated on a 1% agarose gel in 1X SB buffer. The gel was stained with ethidium bromide solution (10 μm/mL), and a single fragment was identified.
ITS analyses
Intraspecific variability among the mono-uredinial isolates was analyzed by characterizing the ITS1 and ITS2 regions of 23 P. pachyrhizi isolates. The combination of the oligonucleotides ITSPP5 (5’-GCAACGGCACTTTACTGGCTC-3′) and ITSPP3 (5’-GTTCAGTGGGTAGTCTCACCTGATT-3′), which were designed by Freire et al. in 2008 and have been successfully used for amplification to assess genetic variability, were used amplify the ITS1, 5.8S and ITS2 regions. PCR reactions were performed with a mixture composed of 1X buffer, 200 μm dNTPs, 1.5 mM MgCl2, 0.25 U Taq DNA Polymerase (Invitrogen), 50 pmol of each oligonucleotide and 50 ng of DNA in a 50 μL reaction volume. The following amplification conditions were used for PCR: one cycle at 94 °C for 5 min; 35 cycles of 94 °C for 60 s, 58 °C for 60 s, 72 °C for 60 s; and 1 cycle at 72 °C for 5 min. To verify the PCR amplification fragments, the products were separated on a 1.0% agarose gel in 1X SB buffer and stained with ethidium bromide (10 μg/mL). The amplified products of approximately 750 bp were purified using the Wizard® SV Gel and PCR Clean-up System (Promega). Cloning was carried out using the pCR®2.1-TOPO cloning vector (Invitrogen®), and plasmid DNA was extracted using a Wizard® Plus Minipreps DNA Purification Systems Kit (Promega). Samples were diluted to a final concentration of 200 μg/μL. Sequencing was performed with Sanger sequencing technology using M13 universal oligonucleotides. Approximately 4–6 clones of the ITS of each isolate were submitted for sequencing.
Data set for ITS analyses
The contigs were obtained with the aid of the Sequencher® program v.4.10. The edited sequences were imported into the program BioEdit 7.0.5.3, and the consensus sequence between the contigs of each isolate was generated by means of visual inspection.
An analysis of molecular variance (AMOVA) using Arlequin software version 3.1 (Excoffier et al., 1992) was used to estimate the partition of genetic variation of the P. pachyrhizi mono-uredinial isolates at one hierarchical level (state of origin). Genetic differentiation was also calculated from AMOVA using the FST index (Weir and Cockerham 1984). Significance estimates were based on 10,000 permutations. In addition, 484 sequences from the ITS region that had previously been deposited in the GenBank database based on the results of Frederick et al. (2002), Barnes et al. (2009), Zhang et al. (2012) and Jorge et al. (2014) were added for the analysis of the global and temporal distribution, as well as to define the ribotype number of P. pachyrhizi isolates. Gene genealogies for the ITS data set were aligned using MEGA7 software and inferred independently using the median-joining (MJ) network method.
(Bandelt et al. 1999) in PopART 1.7 software. For the geographical distribution of the 23 P. pachyrhizi isolates obtained in this study, the ribotypes were plotted manually to reveal their frequencies and spatial relationships.
AFLP analyses
AFLP analysis was carried out according to the methods of Vos et al. (1995), with modifications proposed by Reck et al. (2011). DNA was digested with the EcoRI and MseI restriction enzymes. Following heat inactivation of the restriction endonucleases, the genomic DNA fragments were ligated to adapters, which were then preamplified using the primers E (5’-GACTGCGTACCAATTCA-3′) and M (5’-GATGAGTCCTGAGTAAC-3′). After pre-amplification, selective PCR was performed, in which the selective primers applied were almost identical to the pre-selective E and M, except for specific combinations of two or three nucleotides in the 3′ terminal region. Nine combinations of primers were used: E: ATC + M: GCA; E: TT + M: TT; E: ATC + M: TGT; E: ACA + M: CTG; E: TC + M: AC; E: ACA + M: GCA; E: ACA + M: TC; E: TC + M: AGC; and E: AGC + M: GCA. The amplified fragments were analyzed on a 7% polyacrylamide gel (29:1), and a 10 bp ladder (Invitrogen) was used as a reference.
For diversity analysis, amplification products were manually scored into binary data indicating presence (1), absence (0) or lost plot (9). The software DBOOT v.1.1 (Coelho 2001) was used to evaluate the coefficient of variation (CV%) of the number of amplified markers to determine the reliability parameter of the statistical tests. The number of polymorphic fragments was considered to be optimal when the coefficient of variation was lower than 5%. The software TFPGA 1.3 (Miller, 1997) was used to estimate the allele frequencies for dominant markers (Lynch and Milligan 1994), which included distance and the genetic identity of Nei (1978) (uHe), the proportion of polymorphic loci (P) at the 95% threshold and the variance of the gene frequencies among different locations. AMOVA was also performed as previously described, and the grouping of individuals was performed by the UPGMA (unweighted pair-group method with arithmetic averages), which considers the mean distance values, using the Nei coefficient (1978).
Assessment of disease resistance responses
To evaluate the reaction of the soybean genotypes to the mono-uredinial isolates of P. pachyrhizi obtained in this study, a set of seven soybean accessions were selected based on previous knowledge of the presence of resistance genes to P. pachyrhizi (Rpp) (Table 1) and were tested with 17 isolates. The evaluation of the isolates was carried out through the inoculation and evaluation of detached leaves kept in Petri dishes containing agar-water medium. The third trifoliate leaves of three different plants of each genotype were collected and prepared for culture in plates as described previously.
Frozen spores were used to prepare the suspension of the inoculum containing autoclaved distilled water and 0.01% Tween 20 with a final concentration of 4 × 104 spores/mL. To determine spore viability, the suspension of each isolate was transferred to Petri dishes containing 1% agar-water medium and incubated for 24 h at 21 °C in the dark, and then the germination rate of 100 spores was determined; only isolates with germination rates above 75% were used. The spore suspension was spread homogenously on the lower surface of the leaves using a glass atomizer. Fourteen days after inoculation, the leaves were photographed and evaluated based on quantitative parameters including the lesion sporulation level.
(SL), number of uredinia per lesion (NoU) and frequency of lesions with uredinia (% LU), as described by Yamanaka et al. (2010). Ten lesions per leaf, totaling 90 lesions per genotype (10 lesions × 3 leaves × 3 plants), were analyzed using a stereoscopic magnifying glass. The quantitative data collected were classified into three categories: 1) R, immune or resistant; 2) IM, intermediate; and 3) S, susceptible (Table 2), as proposed by Akamatsu et al. (2013), with modifications.
Results
Isolates
Twenty-three isolates were obtained by means of mono-uredinial culture on detached leaves. Each isolate received a code that refers to its geographical origin (Table 3). Among the isolates obtained, six isolates were from the state of Paraná, five were from the state of Minas Gerais, four were from the state of Goias, four were from Mato Grosso, two were from Rio Grande do Sul and two were from Rondônia.
Throughout the process of obtaining the isolates, the observed symptoms were characteristic of those caused by Asian soybean rust. However, Phakopsora meibomiae (Arth.) Arth and P. pachyrhizi, two species of the genus Phakopsora, cause rust in soybean and occur simultaneously in the same area, producing similar symptoms, but P. pachyrhizi is more aggressive (Ono et al. 1992; Sousa et al. 2007). To detect the specific fungus responsible for soybean rust, the DNA of the isolates was amplified with the Ppa1/Ppa2 primers, resulting in a 332 bp fragment obtained for all the isolates obtained, as described by Frederick et al. (2002), and confirming the identity of the 23 mono-uredinial isolates as belonging to the species P. pachyrhizi (Fig. S1).
Genetic diversity analysis using ITS region data
Amplification of the ITS1 and ITS2 regions with the ITSPP5/ITPP3 primers was performed for the 23 mono-uredinial isolates (GenBank accession numbers MH973780 to MH973802), resulting in a product with approximately 750 bp corresponding to the final part of the 18S rDNA, the complete ITS1–5.8S-ITS2 set and the initial part of the 28S region of P. pachyrhizi. After editing and alignment, the sequences presented a size of 564 bp.
Sequence alignment identified 11 polymorphic sites in the ITS1 region, among which two polymorphic sites presented size variations within a mononucleotide repeat region (polyT), while another site was observed in the dinucleotide repeat region (polyAT). The other polymorphic sites were generated by different substitutions. The isolate L.PG212 presented a change of base (A↔G) at position 39, L.GO412 presented an exchange (G↔A) at position 69, isolate L.CA7A12 presented a change (A↔G) at position 97, L.LD5512 presented a polymorphic site at position 98 (T↔G), and L.LDEB11 presented two polymorphic sites at positions 61 (T↔G) and 91 (T↔A).
Finally, in position 185 of ITS1, an exchange (T↔A) was observed for the isolates L.PF407, L.PG212, L.UB112, L.UB512, L.VL207 and L.GO212 (Fig. 2a). The 5.8S region was highly conserved, and only one polymorphic site at position 287 (A↔G) was observed for the L.LD5511 isolate. Regarding the polymorphisms identified in the ITS2 region, a smaller number was observed, with only seven polymorphic sites, two of which were a size variation in one polyT region. The others were generated by substitutions of bases: seven isolates (L.PF407, L.PG212, L.TAM212, L.LDEB11, L.PL512, L.UB512 and L.GO212) showed an A↔G inversion at position 384. An ↔C inversion was observed in isolates L.LDP211 (at position 456) and L.TAM212 (549), and an A↔G inversion was observed in the L.GO112 (497) isolate. Finally, a T↔A inversion at position 542 was observed for the isolates L.CA7A12, L.PG212, L.TAM212, L.UB112 and L.LD112 (Fig. 2b).

Sequence alignment of the variable sites in P. pachyrhizi isolates. (a) Polymorphisms observed in the ITS1 region and (b) polymorphic sites identified for the ITS2 region. The polymorphic regions are shown. The dots indicate similarity among the sequences, and the dashes indicate “gaps” introduced to compensate for the presence of indels
The 23 isolates defined 11 distinct ribotypes, and the distribution of the localities where they were collected is shown in Fig. 3. Ribotype 1 exhibited the highest frequency, being observed in Conceição das Alagoas (L.CA4A12, L.CA4B12),

Geographic distribution of 23 ITS ribotypes in 13 different regions of Brazil. 1-Conceição das Alagoas-MG; 2-Uberlândia-MG; 3-Londrina-PR; 4-Ponta Grossa-PR; 5-Tamarana-PR; 6-Passo Fundo-RS; 7-Primavera do Leste-MT; 8Rondonópolis-MT; 9-Campo Verde-MT; 10-Lucas do Rio Verde-MT; 11-Vilhena-RO, 12-Goiania-GO and 13-Santo Antônio de Goias-GO
Passo Fundo (L.PF2B07), Lucas do Rio Verde (L.LRV212), Primavera do Leste.
(L.PL512), Vilhena-RO (L.VL107), Campo Verde (L.CV107), Londrina (L.LD112) and.
Rondonópolis (L.RD512). Ribotype 7 was the second most frequent, being found in Passo Fundo (L.PF407), Uberlândia (L.UB112 and L.UB512), Villhena (L.VL207) and Goiânia (L.GO212). All other haplotypes were unique to each locality where they were observed. It was also observed that the same ribotype was found in distinct regions and that the highest number of ribotypes was observed in Londrina. One possible explanation is the higher number of isolates obtained (four) and analyzed from this locality. However, in Mato Grosso state, we observed only the ribotype 1, even we have obtained the same number of isolates, in this case collected from different localities in the state.
We must consider the variability presented by the population collected in Londrina in 2011, which has been maintained in different soybean genotypes under greenhouse conditions, leading to a higher selection pressure and coevolution of hosts and pathogens.
The AMOVA results for the regional analysis showed that 100% of the genetic variation observed was attributed to the differences in the locations where the P. pachyrhizi isolates were collected. From the FST value obtained, it was possible to observe a low genetic differentiation among isolates from the six regions analyzed, demonstrating the lack of spatial genetic structure and that the populations are similar among these locations (Fig. 4).

Analysis of molecular variance (AMOVA) for the ITS region for 23 P. pachyrhizi monouredinial isolates
The analysis spanning 585 ITS sequences from samples collected on five different continents and over four decades (Jorge et al. 2014) plus the samples from this study defined the existence of at least 20 different ribotypes among P. pachyrhizi isolates (Fig. 5). Ribotype 1, the most frequent, was represented by 457 sequences, followed by ribotype 7 with 108 sequences; both ribotypes were represented by samples collected on the five different continents. At the local level, these results reveal a weak spatial and temporal genetic structure for the pathogen, as previously reported (Jorge et al. 2014; Freire et al. 2008); consistent with the AMOVA results, samples from different locations shared the two most frequent ribotypes, including samples collected from 1972 to 1980 (Australia and Asia) (Frederick et al. 2002) that were collected 30 years later, as well as samples obtained in this study. These data suggest a high gene flow among the different regions analyzed.

Network obtained through median-joining for P. pachyrhizi ITS sequences. (a) Network with all available complete ITS sequences for P. pachyrhizi. (b) Network for the ribotypes found in the 13 different regions in Brazil. (c) Network for the ITS regions according to the year of collection. In this analysis, each circle represents a ribotype (identified with numbers) and is colored according to the collection site continent (a) and city (b) and the year of collection (c). The size of the circle within each network is proportional to the number of sequences within the ribotype
However, some unique ribotypes were observed in some samples collected, mostly in the samples from South America R2-R6, R8, R9-R11 (from this study) and R18-R20 (Jorge et a., 2014), but also in materials from North America R13-R15 (Barns et al., 2009) and R16 (Zhang et al. 2012). Furthermore, a single ribotype collected on the African continent R12 (Frederick et al. 2002) was identified (Fig. 5a). With regard to the exclusive ribotypes of South America, nine new ribotypes were identified among the isolates obtained in this study (Fig. 5b).
The temporal network (Fig. 5c) showed that the different ribotypes identified originated mainly after mutations occurred in ribotypes 1 and 7. In addition, the network suggests that the P. pachyrhizi isolates found in Brazil may have originated from multiple independent spore dispersion events from the Asian, African and Oceanian regions (in light gray), as they display ancestral characteristics (represented by the oldest samples, Table S1).
Genotypic diversity analysis with AFLP data
AFLP analysis was performed on 23 P. pachyrhizi isolates using nine sets of primer combinations, and the banding pattern was highly reproducible for 22 isolates. A total of 256 scorable bands were produced, of which 67% were polymorphic. The primer with the highest number of fragments (44) was E: ACA + M: CTG. The primer E: ACA + M: TC had the lowest number of fragments (16). The coefficient of variation from the analysis using the software DBOOT was 3.59%, which is considered reliable for conducting a genetic diversity study (Fig. S2). The average percentage of polymorphic loci was 67%, and Nei’s genetic diversity coefficient was 0.20 (Table 4). The highest genetic diversity was found in Paraná (0.22), and the lowest was detected in Rio Grande do Sul (0.07). Regarding isolates from the same region, the highest similarity was observed between the samples collected in Mato Grosso and Paraná (98%), and the lowest similarity was observed between Minas Gerais and Mato Grosso (94%).
Analysis of molecular variance showed that genetic variance occurred within locations (Fig. 6). When we analyzed samples from different states, the genetic differentiation observed was negative, indicating a low and limited degree of variance among locations, corroborating the data obtained in the ITS analysis.

AMOVA for AFLP markers in 22 P. pachyrhizi samples
The UPGMA dendrogram showed that the total genetic distance among the accessions examined was 50%, revealing two major groups by cluster analysis. Group I contained the majority of the P. pachyrhizi isolates, which were split into subgroups; five were collected in Paraná, four in Minas Gerais, three in Mato Grosso and Goias and two from Rio Grande do Sul and Rondônia. Group II was composed of three isolates from Minas Gerais, Paraná and Goias (Fig. 7). It was not possible to group the isolates exclusively according to their origin, which shows that geographical distances do not correlate with genetic distances among the isolates of P. pachyrhizi analyzed in this study, consistent with the lack of a spatial genetic structure.

Dendrogram generated of the P. pachyrhizi isolates from different regions in Brazil constructed using the paired genetic distances algorithm
Pathogenicity of the P. pachyrhizi isolates from Brazil
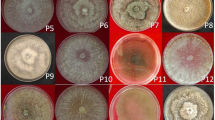
The responses of each of the seven soybean differential genotypes to the 17 P. pachyrhizi mono-uredinial isolates are shown in Table 5 and Fig. 8.

Typical reaction within leaves of differential genotypes 14 days after inoculation with P. pachyrhizi monouredinial isolates. nd not determined
Based on the parameters evaluated, a high variation was observed in the type of reaction between the genotypes and the isolates analyzed. Both extremes, as well as intermediate levels, were observed for all three parameters evaluated. The isolates L.LDEB11, L.PL512, L.PF2B07, and L.VL207 exhibited the same pathogenicity profile in seven differentials. These isolates came from different locations, showing that there is no association between the pathogenic profile and the geographical origin of the isolates. The isolates L.LD112 and L.GO212 were the most pathogenic among the evaluated isolates, causing disease or a susceptibility reaction in five of the seven genotypes tested, three of which have a locus of resistance to Asian soybean rust.
(Rpp1, Rpp2 and Rpp6). On the other hand, the isolates L.CV107 and L.VL107, both collected in the year 2007, were the least pathogenic, containing avirulence factors recognized by many of the Rpp genes present in the soybean differentials.
Phenotypic analysis carried out on detached leaves showed that PI 587855 (Rpp1b) was susceptible to all isolates tested, showing the same response profile as the susceptible control, BRS 184. We also observed a high number of susceptible reactions in PI 200492 (Rpp1), with 10 incompatible reactions. On the other hand, PI.
230,970 (Rpp2) presented the highest number of resistance interactions (nine out of 17 tested), followed by the Rpp4 genotype, which presented seven incompatible interactions (52.9%), but no susceptible reactions, thus it was the most effective gene against the Brazilian isolates tested in this study. PI 200487 (Rpp5) also presented many cases of resistance reactions among the total of eleven interactions tested (54.5%), but at least one isolate, collected in Goias in 2012, was able to overcome the resistance conferred by this gene. The other genotypes presented variable reactions to the tested isolates (Figs. 8 and 9).

Frequency of infection response types observed in the seven soybean differentials after inoculation with P. pachyrhizi monouredinial isolates
These response profiles reflected the average for each evaluated parameter. Rpp5 mediated the most cases (six) of low SL, including SL values of 0.03 and 0.21 for L.VL0107 and L.VL207, respectively, and Rpp2 mediated low SL values for five isolates, including values of 0.13 for L.VL0107 and 0.2 for L.UB112. Rpp4 presented four cases with low SL, which was expected as this gene presented the largest number of intermediate reactions. The number of uredinia per lesion (NoU) showed the same pattern as that observed for the SL parameter.
The pathogenicity profiles of the isolates showed different responses to the seven genotypes tested, indicating that there are different virulence factors among the P. pachyrhizi isolates found in the soybean fields of Brazil and that the 17 isolates analyzed represent 15 different pathotypes or races of the pathogen. Comparatively, the variability in the virulence of the isolates was greater than the molecular variability.
Discussion
The specific detection of soybean rust fungus for the 23 isolates analyzed in this study demonstrates the high frequency and wide distribution of P. pachyrhizi in soybean-producing fields in Brazil, corroborating results reported in other studies (Sousa et al. 2007; Freire et al. 2008). These results do not exclude the presence of P. meibomiae in Brazilian soybean fields, but they indicate that the incidence of this species may be extremely low compared to the occurrence of P. pachyrhizi.
The results obtained in the present work demonstrated that analysis of the ITS regions was effective for the identification of intraspecific variation among mono-uredinial isolates of P. pachyrhizi, revealing the presence of at least 11 different ribotypes in Brazil based on our dataset. Likewise, the results also showed the effectiveness of AFLP markers in assessing the genetic diversity of this pathogen since a large number of markers and a high degree of polymorphism were observed, revealing the existence of high intraspecific variability in the collection of P. pachyrhizi isolates obtained, as already observed in other studies (Rocha et al. 2015).
Although the variability observed at the population level is expected to be greater than found in at the isolate level, the data obtained in the present study was still effective and informative, allowing the identification of rare types in a population (as demonstrated by the number of single ribotypes observed in this study), as well as the analysis of their correlations at the race level.
The analysis of the ribotype distribution among the different collection sites and the phylogenetic analysis using AFLP markers and ITS region polymorphisms demonstrated a diverse distribution of the isolates with considerable genetic variability. A high level of sequence variation in the ITS region and AFLP markers has been documented in other studies on P. pachyrhizi populations (Freire et al. 2008; Zhang et al. 2012; Jorge et al. 2014; Rocha et al. 2015). This variability can be explained by the high evolutionary potential present in rust species, especially P. pachyrhizi, which could be related to its population size; a high number of mutations can occur in each generation, considering the number of spores that can emerge from one unique single lesion. Additionally, a sexual cycle has not yet been described for this pathogen, so this variability could also be derived from somatic recombination that might combine nuclei originating from different hyphae, especially considering that hyphae anastomoses have already been described (Vittal et al. 2012). Finally, the complex genome, large size and high level of repetition derived from retrotransposons.
(Loehrer et al. 2014) can support many types of recombination.
Although a high diversity level was observed, the AMOVA results revealed a weak, or even a lack, of genetic structure in P. pachyrhizi populations, corroborating previous studies that also used ITS region polymorphisms and AFLP markers to analyze the diversity and genetic structure in populations collected on the American continents (Brazil, Argentina, Paraguay and United States of America) from 2003 to.
2009 (Rocha et al. 2015; Jorge et al. 2014; Zhang et al. 2012; Freire et al. 2008). Similar results were also observed in samples collected in four agro-ecological zones in Nigeria in 2005, that were tested for SSR markers (Twizeyimana et al. 2011). These data suggest high rates of gene flow among P. pachyrhizi populations, which can be explained by the dissemination of soybean rust that occurs mainly through spore dispersion by the wind to nearby crops or over long distances (Embrapa, 2004; Yorinori et al. 2004), favoring isolate distribution and rapid homogenization into different geographic areas.
Regarding the temporal distribution, the two most frequent ribotypes, 1 and 7, which also showed the most widespread geographic distribution among the ITS ribotypes, may also have an ancient origin. These ribotypes were also found in isolates collected in Australia (1972 and 1979), Hawaii (1995 and 1998), India (1973),
Indonesia (1972), Philippines (1977), Taiwan (1972 and 1980), Thailand (1976) and Zimbabwe (2000), suggesting the occurrence of long-distance dispersion among these different areas, reinforcing the hypothesis of a global migration of P. pachyrhizi isolates from Asia, Australia and Africa and corroborating the results obtained in other studies (Pan et al. 2006; Zhang et al. 2012).
Finally, although a global distribution has been observed, some ribotypes appear to be present only in Africa, North America and South America, which can be explained by the occurrence of mutation events and the introduction of genetically distinct isolates. Further studies are necessary to determine whether these rare or specific haplotypes reflect locally adapted genotypes.
Based on the responses of the soybean differentials, the mono-uredinial isolates described in this study presented different response profiles in the seven genotypes used, indicating the existence of different avirulence factors among P. pachyrhizi isolates found in Brazil, and indicating that the 17 isolates tested might represent 16 different pathotypes or races of the pathogen. L.LD112 and L.GO212 showed a highly virulent profile, which could represent a threat since these isolates were able to overcome the resistance of most of the Rpp genes tested.
Pathogenic diversity was observed in Brazil shortly after Asian soybean rust was first identified in the country (in 2002), when none of the four dominant resistance genes described in that time, Rpp1-Rpp4, were effective against the pathotypes that occurred in 2003 (Yorinori et al. 2004). This variability has still been observed in more recent studies, that have analyzed samples from Brazilian soybean-producing areas (Yamanaka et al. 2010; Akamatsu et al. 2013, 2017) and can be explained, among other factors already cited, by the tropical and subtropical climate of this continent and the lack of rigorous winters, which allows the continuous presence of a host. The simultaneous occurrence of soybean and alternative hosts contributes to the maintenance of the life cycle of P. pachyrhizi, playing an important role in promoting the diversity presented by the fungus in these regions (Akamatsu et al. 2013).
Our data revealed PI 230970 (Rpp2), PI 459025 (Rpp4) and PI 200487 (Rpp5) as the most effective sources of resistance to isolates found in Brazil. Yamanaka et al. (Yamanaka et al. 2010 and 2011) also observed the resistance conferred by these genotypes during the selection of material with resistance to soybean rust upon inoculation with spore populations collected in Japan (JRP) and Brazil (BRP-2). Lemos et al. (2011) evaluated the effects of Rpp2, Rpp4 and Rpp5 on the NO6–12 genotype, a pyramid containing the three Rpp genes, and observed a higher contribution of Rpp5 to the same parameters used in this study (SL, NoU and %LU).
Rpp2 and Rpp4 resistance responses were also observed after inoculation with P. pachyrhizi populations collected in Argentina, Paraguay and Brazil from 2007 to 2015 (Akamatsu et al. 2013, 2017). Although the same parameters were not used for evaluation, these genes were also effective when the material was infected with isolates collected in Taiwan, Philippines, Australia, Hawaii, South Africa and.
Paraguay (Rpp2) or Taiwan, Philippines, India, Hawaii, Thailand, Brazil and Paraguay (Rpp4) (Bonde et al. 2006).
As previously reported (Bonde et al. 2006; Yorinori et al. 2008; Pham et al. 2009; Akamatsu et al. 2013, 2017), the resistance mediated by PI 200492 was ineffective against most of the Brazilian isolates tested in this study; resistance was only observed for the L.STA212 isolate, and no immune interaction was observed.
Most of the isolates evaluated for pathogenicity were defined as distinct ribotypes in the ITS region analysis. The isolates L.PG212, L.LDEB11, L.GO412, L.CA7A12, L.LDP211, L.GO112 and L.TAM212 showed single nucleotide polymorphisms (SNPs) unique to each ITS sequence. As proposed by Zhang et al. (2012), these SNPs can be used as molecular markers for the detection and identification of specific P. pachyrhizi races or pathotypes. Comparatively, the diversity revealed in the virulence of the isolates was greater than the genetic.
diversity.
Twizeyimana et al. (2011), in an analysis of 116 P. pachyrhizi isolates collected in Nigeria using 18 microsatellite markers, also observed a low correlation between the pathogenic and genetic variation data. According to Bux et al. (2012), genetic diversity, specially based on local regions of the genome, as the ITS regions, is independent of pathogenicity because the entire pathogen genome evolves faster than pathogenicity-regulating genes. Thus, a direct relationship between pathogenicity and genetic data would be observed if pathogenicity were controlled by many genes distributed throughout the genome.
Knowledge about the variability and genetic structure of P. pachyrhizi populations is extremely important, as such knowledge enables the design of better strategies for pathogen control. In the future, the new sequencing technologies might offer an incomparable possibility to access the global variability of this specie at the genome level revealing questions about its genome plasticity and evolution.
References
Akamatsu, H., Yamanaka, N., Yamaoka, Y., Soares, R. M., Morel, W., Ivanonich, A. J. G., Bogado, A. N., Kato, M., Yorinori, J. T., & Suenaga, K. (2013). Pathogenic diversity of soybean rust in Argentina, Brazil, and Paraguay. Journal of General Plant Pathology, 79, 28–40.
Akamatsu, H., Yamanaka, N., Soares, R. F., Ivancovich, A. J. G., Lavilla, M. A., Bogado, A. N., Morel, G., Scholz, R., Yamaoka, Y., & Kato, M. (2017). Pathogenic variation of south American Phakopsora pachyrhizi populations isolated from soybeans from 2010 to 2015. Japan Agricultural Research Quarterly, 51, 221–232.
Bandelt, H. J., Forster, P., & Rohl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution, 16, 37–48.
Barnes, C. W., Szabo, L. J., & Bowersox, V. C. (2009). Identifying and quantifying Phakopsora pachyrhizi spores in rain. Phytopathology, 99, 328–338.
Bonde, M. R., Nester, S. E., Austin, C. N., Stone, C. L., Frederick, R. D., Hartman, G. L., & Miles, M. R. (2006). Evaluation of virulence of Phakopsora pachyrhizi and P. meibomiae isolates. Plant Disease, 90, 708–716.
Bromfield, K. R., & Hartwig, E. E. (1980). Resistance to soybean rust and mode of inheritance. Crop Science, 20, 254–255.
Bux, H., Rasheed, A., Mangrio, S. M., Abro, A. S., Shah, S. J. A., Ashraf, M., & Chen, S. (2012). Comparative virulence and molecular diversity of stripe rust (Puccinia striiformis f.sp. tritici) collections from Pakistan and United States. International Journal of Agriculture and Biology, 14, 851–860.
Childs, S. P., King, Z. R., Walker, D. R., Harris, D. K., Pedley, K. F., Buck, J. W., Boerma, H. R., & Li, Z. (2018). Discovery of a seventh Rpp soybean rust resistance locus in soybean accession PI605823. Theoretical Applied Genetics, 131, 121–141.
Coelho, A. S. G. (2001). Software: Dboot—Avaliação de dendogramas baseados em estimativas de distâncias/similaridades genéticas através do procedimento de bootstrap, versão 3.0. Goiânia: Universidade Federal de Goias.
Dellaporta, S. L., Wood, J., & Hicks, J. B. (1983). A plant minipreparation: Version II. Plant Molecular Biology Reporter, 1, 19–21.
Frederick, R. D., Synder, C. L., Peterson, G. L., & Bonde, M. R. (2002). Polymerase chain reaction assays for the detection and discrimination of soybean rust pathogens Phakopsora pachyrhizi and P. meibomiae. Phytopathology, 92, 217–227.
Freire, M. C. M., Oliveira, L. O., Almeida, A. M. R., Schuster, I., Moreira, M. A., Liebenberg, M. M., & Mienie, C. M. S. (2008). Evolutionary history of Phakopsora pachyrhizi (the Asian soybean rust) in Brazil based on nucleotide sequences of the internal transcribed spacer region of the nuclear ribosomal DNA. Genetics and Molecular Biology, 31, 920–931.
Freire, M. C. M., Silva, M. R., Zhang, X., Almeida, A. M. R., Stacey, G., Oliveira, L. O. (2012). Nucleotide polymorphism in the 5.8S nrDNA gene and internal transcribed spacers in Phakopsora pachyrhizi viewed from structural models. Fungal Genetics and Biology, 49:95–100.
Garcia, A., Calvo, E. S., Souza Kiihl, R. A., Harada, A., Hiromoto, D. M., & Vieira, L. G. (2008). Molecular mapping of soybean rust (Phakopsora pachyrhizi) resistance genes: Discovery of a novel locus and alleles. Theoretical and Applied Genetics, 117, 545–553.
Hartman, G. L., Miles, M. R., & Frederick, R. D. (2005). Breeding for resistance to soybean rust. Plant Disease, 89, 664–666.
Hartwig, E. E. (1986). Identification of a fourth major gene conferring resistance to soybean rust. Crop Science, 26, 1135–1136.
Hartwig, E. E., & Bromfield, K. R. (1983). Relationships among three genes conferring specific resistance to rust in soybeans. Crop Science, 23, 237–239.
Jorge, V. R., Silva, M. R., Guillin, E. A., Freire, M. C. M., Schuster, I., Almeida, A. M. R., & Oliveira, L. O. (2014). The origin and genetic diversity of the causal agent of Asian soybean rust, Phakopsora pachyrhizi, in South America. Plant Pathology, 64, 729–737.
Langenbach, C., Schultheiss, H., Rosendahl, M., Tresch, N., Conrath, U., & Goellner, K. (2015). Interspecies gene transfer provides soybean resistance to a fungal pathogen. Plant Biotechnology Journal, 14, 699–708.
Li, S., Smith, J. R., Ray, J. D., & Frederick, R. D. (2012). Identification of a new soybean rust resistance gene in PI567102B. Theoretical Applied Genetics, 125, 133–142.
Lemos, N. G., Braccini, A. L., Abdelnoor, R. V., Oliveira, M. C. N., Suenaga, K., & Yamanaka, N. (2011). Characterization of genes Rpp2, Rpp4, and Rpp5 for resistance to soybean rust. Euphytica, 182, 53–64.
Lynch, M., & Milligan, B. G. (1994). Analysis of population structure with RAPD markers. Molecular Ecology, 3, 91–99.
Loehrer, M., Vogel, A., Huettel, B., Reinhardt, R., Benes, V., Duplessis, S., Usadel, B., & Schaffrath, U. (2014). On the current status of Phakopsora pachyrhizi genome sequencing. Frontiers in Plant Science, 5, 377.
Martin, K. J. (2007). Introduction to molecular analysis of ectomycorrhizal communities. Soil Science Society America Journal, 71, 601–610.
Mclean, R. J., & Byth, D. E. (1980). Inheritance of resistance to rust (Phakopsora pachyrhizi) in soybeans. Australian Journal of Agricultural Research, 31, 951–956.
Meudt, H. M., & Clarke, A. C. (2007). Almost forgotten or latest practice? AFLP applications, analysis and advances. Trends Plant Science, 12, 106–117.
Miles, M. R., Bonde, M. R., Nester, S. E., Berner, D. K., & Frederick, R. D. (2011). Characterizing resistance to Phakopsora pachyrhizi in soybean. Plant Disease, 95, 577–581.
Ono, Y., Buritica, P., & Hennen, J. F. (1992). Delimitation of Phakopsora, Physopella and Cerotelium and their species on Leguminosae. Mycological Research, 96, 825–850.
Pham, T. A., Miles, M. E., Frederick, R. D., Hill, C. B., & Hartman, G. L. (2009). Differential responses of resistant soybean entries to isolates of Phakopsora pachyrhizi. Plant Disease, 93, 224–228.
Reck, M., Benício, L. M., Ruas, E. A., Rodrigues, L. A., Ruas, P. M., Ortiz, M. A., Talavera, S., Urtubey, E., Stuessy, T., & Weiss-Schneeweiss, H. (2011). Karyotype and AFLP data reveal the phylogenetic position of the Brazilian endemic Hypochaeris catharinensis (Asteraceae). Plant Systematics and Evolution, 296, 231–243.
Rocha, C. M. L., Vellice, G. R., García, M. G., Pardo, E. M., Racedo, J., Perera, M. F., de Lucía, A., Gilli, J., Bogado, N., Bonnecarrere, V., German, S., Marcelino, F., Ledesma, F., Reznikov, S., Ploper, L. D., Welin, B., & Castagnaro, A. P. (2015). Use of AFLP markers to estimate molecular diversity of Phakopsora pachyrhizi. Electronic Journal of Biotechnology, 18, 439–444.
Sousa, P. F. C., Alves, E., Castro, H. A. C., Souza, P. E., Almeida, A. M. R., & Hochenbach, S. R. (2007). Distribuição e identificação de Phakopsora pachyrhizi, agente causal da ferrugem da soja no Estado de Minas Gerais. Ciência e Agrotecnologia, 31, 672–677.
Twizeyimana, M., & Hartman, G. L. (2010). Culturing Phakopsora pachyrhizi on detached leaves and urediniospore survival at different temperatures and relative humidities. Plant Disease, 94, 1453–1460.
Twizeyimana, M., Ojiambo, P. S., Haudenshield, J. S., Caetano-Anollés, G., Pedley, K. F., Bandyopadhayay, R., & Hartman, G. L. (2011). Genetic structure and diversity of Phakopsora pachyrhizi isolates from soyabean. Plant Pathology, 60, 719729.
Vittal, R., Yang, H. C., & Hartman, G. L. (2012). Anastomosis of germ tubes and migration of nuclei in germ tube networks of the soybean rust pathogen, Phakopsora pachyrhizi. European Journal of Plant Pathology, 132, 163–167.
Vos, P., Hogers, R., Bleeker, M., Reijans, M., Van De Lee, T., Hornes, M., Frijters, A., Pot, J., Peleman, J., & Kuiper, M. (1995). AFLP: A new concept for DNA fingerprinting. Nucleic Acids Research, 23, 4407–4414.
Weir, B. S., & Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution, 38, 1358–1370.
Yamanaka, N., Yamaoka, Y., Kato, M., Lemos, N. G., Passianotto, A. L. L., Santos, J. V. M., Benitez, E. R., Abdelnoor, R. V., Soares, R. M., & Suenaga, K. (2010). Development of classification criteria for resistance to soybean rust and differences in virulence among Japanese and Brazilian rust populations. Tropical Plant Pathology, 35, 153–162.
Yamanaka, N., Lemos, N. G., Akamatsu, H., Yamaoka, Y., Silva, D. C. G., Passianoto, A. L. L., Abdelnoor, R. V., Soares, R. M., & Suenaga, K. (2011). Soybean breeding materials useful for resistance to soybean rust in Brazil. Crop Science, 45, 385–395.
Yamaoka, Y., Fujiwara, I., Kakishima, M., Katsuya, K., Yamada, K., & Hagiwara, H. (2002). Pathogenic races of Phakopsora pachyrhizi on soybean and wild host plants collected in Japan. Journal of General Plant Pathology, 68, 52–56.
Yamanaka, N., Morishista, M., Mori, T., Muraki, Y., Hasegawa, M., Hossain M. M., Yamaoka, Y., Kato, M. (2016). The locus for resistance to Asian soybean rust in PI587855. Plant Breeding, 135, 21–626.
Yorinori, J. T. (2008). Soybean germplasm with resistance and tolerance to Asian rust and screening methods. JIRCAS Working Report, 58, 70–70.
Yorinori, J. T., Paiva, W. M., Frederick, R. D., Costamilan, L. M., Bertagnolli, P. F. H., Godoy, G. E., Nunes, C. V., & Jr, J. (2005). Epidemics of soybean rust (Phakopsora pachyrhizi) in Brazil and Paraguay from 2001 to 2003. Plant Disease, 89, 675–677.
Yorinori, J. T., Junior, J. N., & Lazzarotto, J. J. (2004). Ferrugem “asiática” da soja no Brasil: evolução, importância, economia e controle (Série Documentos, 247) (p. 36). Londrina: Embrapa.
Zhang, X. X., Freire, M. C. M., Le, M. H., Oliveira, L. O., Pitkin, J. W., Segres, G., Concibido, V. C., Baley, G. J., Hartman, G. L., Upchurch, G., Pedley, K. F., & Stacey, G. (2012). Genetic diversity and origins of Phakopsora pachyrhizi isolates in the United States. Asian Journal of Plant Pathology, 6, 52–65.
Acknowledgments
The authors would like to express their gratitude to all who provided assistance in collecting the material and conducting all the experiments. We greatly appreciate the financial support of CAPES, CNPq and Embrapa for this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest that could be perceived as prejudicial to the impartiality of the reported research.
Ethical statement
This manuscript has not been submitted to any other journal for simultaneous consideration. All the co-authors consented to the manuscript submission and have contributed sufficiently to the scientific work and therefore share collective responsibility and accountability for it. This work does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
ESM 1
(XLSX 30 kb)
Rights and permissions
About this article

Cite this article
Darben, L.M., Yokoyama, A., Castanho, F.M. et al. Characterization of genetic diversity and pathogenicity of Phakopsora pachyrhizi mono-uredinial isolates collected in Brazil. Eur J Plant Pathol 156, 355–372 (2020). https://doi.org/10.1007/s10658-019-01872-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-019-01872-2