
Abstract
Ceratocystis pirilliformis was first described from wounds on Eucalyptus trees in Australia and subsequently found as a common wound inhabitant on these trees in South Africa. During a recent disease survey carried out to identify Ceratocystis species infecting tree wounds in South Africa, C. pirilliformis was isolated from various tree species, including a native Rapanea species and two non-native hosts, Acacia mearnsii and E. grandis growing in the Western Cape Province. These new collections from the Western Cape Province, together with isolates of C. pirilliformis previously collected from Eucalyptus species in the eastern part of the country, provided the opportunity to investigate the possible movement of the pathogen since it was first recorded in South Africa. The identity of newly collected isolates of C. pirilliformis was confirmed using DNA sequence comparisons using partial β-tubulin and TEF-1α gene regions. Microsatellite markers were screened on a collection of C. pirilliformis isolates from the Western Cape Province. Results showed that this population has higher levels of population genetic estimates such as private allele frequency, allelic richness, and gene diversity. Inoculation tests were performed to determine whether C. pirilliformis is a pathogen of A. mearnsii, which is planted as an important plantation forestry tree in the country. All isolates were shown to be highly virulent. Results of the study consequently revealed that C. pirilliformis has a wider host range and geographic distribution in South Africa than previously recognized and that it is a potentially important pathogen of A. mearnsii.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Species of Ceratocystis include numerous economically important fungal pathogens that cause diseases of a wide range of plants, especially angiosperm trees (Kile 1993; Roux and Wingfield 2013; Seifert et al. 2013). Disease symptoms include rots of various root crops, cankers on the stems of trees, streaked discolouration of the xylem tissues, wilt, and tree death (Kile 1993; Roux and Wingfield 2009). Ceratocystis species require wounds to infect hosts that arise from natural or accidental damage to roots or stems (DeVay et al. 1968; Roux et al. 2007). Some species have also established important symbioses with wood-infesting insects that facilitate their dispersal (Heath et al. 2009; Al Adawi et al. 2013; Ploetz et al. 2013; Soulioti et al. 2015).
Since the first report of a disease caused by a species of Ceratocystis on exotic plantation trees in Brazil (Ribeiro et al. 1988), there have been increasing numbers of studies reporting new Ceratocystis diseases on forest plantation trees globally (Roux and Wingfield 2009; Tarigan et al. 2011). These include the Ceratocystis wilt disease of Acacia mearnsii De Wild. caused by C. albifundus M.J. Wingf., De Beer & M.J. Morris in South Africa (Morris et al. 1993; Wingfield et al. 1996), wilt and death of Eucalyptus species caused by C. eucalypticola M. van Wyk & M.J. Wingf. (as C. fimbriata sensu lato) in Brazil (Ferreira et al. 1999; van Wyk et al. 2012), the Republic of Congo (Roux et al. 2000), Uganda (Roux et al. 2001) and Uruguay (Barnes et al. 2003a) and a serious canker and wilt disease of A. mangium Willd. in Indonesia caused by C. manginecans M. van Wyk, Al Adawi & M.J. Wingf. (Tarigan et al. 2011; Fourie et al. 2015).
Until recently, the genus Ceratocystis was used in the broad sense to include a wide variety of morphologically and ecologically similar, but in many cases very different, fungi. These have now been accommodated in a suite of different genera as suggested by Wingfield et al. (2013a) and implemented by de Beer et al. (2014). Ceratocystis is now restricted to species phylogenetically related to Ceratocystis fimbriata sensu stricto, first described for the sweet potato pathogen by Halsted (1890). The name Ceratocystis used in the present study is based on the classification provided by de Beer et al. (2014).
Ceratocystis pirilliformis I. Barnes & M. J. Wingf. was first discovered as the causal agent of sap-stain on wounded Eucalyptus nitens (H. Deane & Maiden) Maiden near Canberra in Australia (Barnes et al. 2003b). It was subsequently found on Eucalyptus species in the Mpumalanga and KwaZulu-Natal Provinces of South Africa (Roux et al. 2004). Based on low levels of genetic diversity in the country, Nkuekam et al. (2009) suggested that C. pirilliformis was most likely introduced to South Africa from Australia. This view, along with the fact that C. pirilliformis was first found in Australia (Barnes et al. 2003b), is further supported by the absence of any C. pirilliformis-associated diseases on E. nitens in Australia where this tree is native (Roux et al. 2004).
During recent tree disease surveys in the Western Cape Province of South Africa, a Ceratocystis species with pear-shaped ascomatal bases, typical of those of C. pirilliformis, was consistently found colonizing artificially-induced wounds on Acacia mearnsii in the Stellenbosch area. It was also found on the stumps of recently felled A. mearnsii, E. nitens and Rapanea species in the Garden Route National Park of the Western Cape Province. This raised questions concerning the movement of C. pirilliformis in South Africa subsequent to its first discovery in the eastern part of the country. The aims of this study were thus: i) to confirm the identification of the newly collected isolates resembling C. pirilliformis from the Western Cape areas, ii) to trace the movement of the pathogen in South Africa using microsatellite markers, and iii) to determine whether the fungus has the capacity to infect A. mearnsii by conducting pathogenicity trials using selected isolates collected in this study.
Materials and methods
Fungal isolates
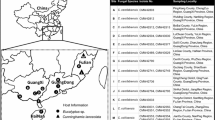
Collections of Ceratocystis species were made from A. mearnsii in the Stellenbosch area (ST), and A. mearnsii, Eucalyptus grandis W. Hill ex Maid. and Rapanea species in the Garden Route National Park (GRNP) of the Western Cape Province (WC) (Table 1, 1S). Wounds on the stems of trees that had either been artificially induced with a machete or axe, or arose from tree harvesting, were surveyed 4 weeks after wounding in December 2013, and January 2014. Bark flaps surrounding the wounds were examined for sexual structures typical of Ceratocystis species using a 10 × magnification hand lens. Pieces of exposed bark bearing ascomata were collected separately for each tree in paper bags, and transported to a laboratory in order to make isolations. Samples were inspected regularly for the presence of Ceratocystis species producing ascospore masses at the tips of ascomatal necks. Cultures were made by directly transferring single drops of ascospores to separate Petri dishes (65 mm) containing 2 % MEA (20 g Malt Extract; 20 g Agar, Biolad, Midland, South Africa) supplemented with 100 mg/l of the antibiotic streptomycin (SIGMA-ALDRICH, Steinheim, Germany) and incubated at 25 °C for 2 weeks in the dark. All isolates obtained in this study were deposited in the culture collection (CMW) of the Forestry and Agricultural Biotechnology Institute (FABI), University of Pretoria, South Africa (Table 1S).
In addition to the isolates obtained in this study, isolates of C. pirilliformis from the study of Nkuekam et al. (2009) were obtained from the CMW collection in FABI and used for comparative purposes when considering the genetic diversity of populations of C. pirilliformis in South Africa. These isolates had been collected from Bushbuckridge (Waterhoutboom) and Sabie (Bergvliet Plantation) in the Mpumalanga Province and Paulpietersburg (Eersteling Plantation) in the Province of KwaZulu-Natal (ESA), South Africa (Table 1, 1S).
DNA extraction
Genomic DNA was extracted from all the isolates collected in the survey and those obtained from the CMW culture collection. Mycelium was scraped from the surfaces of the cultures using a sterile surgical scalpel and transferred to 1.5 ml Eppendorf tubes. Genomic DNA was extracted following the technique described by Möller et al. (1992). The concentration of the extracted DNA was quantified using a ND-1000 spectrophotometer (NanoDrop Technologies, Inc, USA), and was diluted to a working concentration of 15 ng/μl. After quantification, DNA extracts were used in subsequent PCR reactions for species identification and for microsatellite amplification.
Identification of fungi
Isolates (Table 1, 1S) collected from ST and GRNP were identified based on morphological characters and DNA sequence comparisons. All isolates were identified using light microscopy (C-DS, SMZ645, NIKON, Japan) following the description of Barnes et al. (2003b). In addition to the morphological identifications, two isolates (CMW42018, CMW42020) from ST and two isolates (CMW42042, CMW42336) from GRNP were randomly selected and used for species identification using DNA sequence comparisons.
DNA sequence analysis was performed following the techniques described by Lee et al. (2015) based on part of the β-tubulin 1 gene region (BT1) using primers Bt1a and Bt1b (Glass and Donaldson 1995), and the Translation Elongation Factor-1 alpha (TEF-1α) regions with primers TEF1F and TEF2R (Jacobs et al. 2004). The PCR amplification was carried out in a total volume of 15 μl containing 15 ng/μl of genomic DNA, 0.5 μl of each forward and reverse primer (10 pM) for both BT1 and TEF-1α gene regions, 5 μl of 5 × reaction buffer containing 5 mM dNTPs and 15 mM MgCl2 (Bioline in London, UK) and 0.09 μl of MyTaq™ DNA polymerase (Bioline in London, UK). PCR reactions were conducted in a Veriti™ 96 well Thermal Cycler (Applied Biosystem™, Foster City, CA) using the same conditions as those described by van Wyk et al. (2006). The amplification products stained with GelRed™ (Biotium Incorporation, USA) nucleic acid dye on a 1.5 % (w/v) agarose gel in 1 × TAE buffer (40 mM Tris, 20 mM acetic acid, and 1 mM EDTA at pH 8.0) were separated by electrophoresis (Mini-Sub® Cell CT, Biolad, USA), and then visualized under UV light (Gel Doc™ EZ Imager, Bio-Rad, Richmond, CA).
Phylogenetic analyses
Sequences for each gene region were combined in a single data set and were aligned with published sequences of Ceratocystis species retrieved from GenBank (Table 2S), using the online version of MAFFT ver.7.215 (http://mafft.cbrc.jp/alignment/server/) (Katoh et al. 2002). Maximum likelihood (ML) analyses were performed using RAxML HPC BlackBox ver.8.1.11 (Stamatakis 2006; Stamatakis et al. 2008) using the default option with GTR substitution model implemented in the CIPRES cluster server (https://www.phylo.org/) at the SanDiego Supercomputing Center.
Pathogenicity tests
The ability of Ceratocystis species collected in this study to cause disease on A. mearnsii was tested on approximately 3-year-old A. mearnsii trees following the same technique as previously described by Lee et al. (2015). Twenty trees were inoculated with each isolate (100 trees in total) and 20 trees inoculated with sterile agar were used as controls. Inoculations were conducted in a jungle stand near Pretoria East, Gauteng Province of South Africa in December 2014. Five isolates (CMW42042, CMW42336, CMW42018, CMW42019, CMW42020) were used in the inoculation experiment and lesion lengths were measured 6 weeks after the inoculation. Analysis of Variance (ANOVA) and Tukey’s Honestly Significance Difference (Tukey’s HSD) test were used to determine whether there were significant differences in aggressiveness between isolates used for the inoculation trial based on a P-value computed using R ver.3.1.0 (http://www.r-project.org/; R Core Team 2014). Re-isolations were made to ensure that the inoculated fungi were responsible for the infections observed and to fulfil the requirements of Koch’s postulates.
Microsatellite amplification and scoring of alleles
To determine the genetic diversity and contemporary population structure of the isolates, 17 microsatellite primer sets (Table 3S), previously developed by Barnes et al. (2001) and Steimel et al. (2004), were screened using the same conditions described by the authors, with slight modifications in the annealing temperature (Table 3S).
The size of microsatellite amplicons was determined with an ABI PRISM™ 3500xl POP 7™ Automated DNA sequencer (Applied Biosystcms™ / Life Technologies. Carlsbad, US). GENESCAN-600 LIZ (Applied Biosystcms™ / Life Technologies. Carlsbad, US) was used as the internal size standard. Determination of expected allele sizes, based on fragment length, was accomplished using GENEMARKER ver.2.2.0 (Softgenetcis, LLC, USA).
Each allele was sequenced to confirm the size of PCR products amplified with each microsatellite marker. Each confirmed product length was treated as a unique allele. PCR reaction conditions and DNA sequencing of these alleles followed the same procedures described above.
Population genetic analyses and random mating
To determine the movement of C. pirilliformis in South Africa, isolates were grouped into three larger populations based on sampling location: ST from Stellenbosch area, GRNP from the Garden Route National Park and ESA from the KwaZulu-Natal and Mpumalanga Provinces. The confidence for being able to cluster each sampling site into these populations was based on results from Rst calculations (Slatkin 1995) implemented in ARLEQUIN ver.3.5.2.1 (Excoffier et al. 2005; Excoffier and Lischer 2010). A Multi-Locus Haplotype (MLH) data set, in which genetically identical haplotypes were clone-corrected from each of the locations, was generated for each population and used for further analysis. The number of different alleles (Na), the number of effective alleles (Ne; Nielsen et al. 2003) and gene diversity (H; Nei 1973) were calculated using the software program GENALEX ver.6.5 (Peakall and Smouse 2012). ADZE ver.1.0 (Szpiech et al. 2008) was used to compute the private allelic richness (P) and allelic richness (AR). Given that populations had different sample sizes in this study, a rarefaction procedure using ADZE, capable of adjusting P and AR to a specific sample size, was used. The sample sizes were standardized to the smallest clone-corrected population, GRNP in this study (N = 7). R package “poppr” (Kamvar et al. 2014) was used to compute the genotypic richness (eMLG; Hurlbert 1971) and three measures of genotypic diversity using the non-clone-corrected data set. These genotypic estimates computed included the Shannon-Weiner Index (G; Shannon 2001), Stoddart and Taylor’s Index (GD; Stoddart and Taylor 1988) and unbiased genotypic diversity (Hexp) where genotype frequencies were standardized according to sample size (Nei 1978).
Evidence for random mating for each C. pirilliformis population was tested using the R package “poppr” with 999 randomizations (Kamvar et al. 2014). The null hypothesis of no linkage observed amongst alleles at different loci would be indicative of the population having undergone sexual reproduction based on the index of association (I A ) and multilocus linkage disequilibrium (r‐D).
Population genetic structure
Genetic structure for the studied populations was inferred based on the Bayesian genotype clustering method implemented in STRUCTURE ver.2.3.4 (Pritchard et al. 2000). The optimum K value, referred to as the optimal number of inferred genetic groups, was determined with the admixture model with correlated allele frequency. The ‘K’ value was set to test from 1 to 9 possible clusters with 30 iterations, and the program was simulated with 5,000,000 runs with the burn-in period set at 500,000 runs. Results obtained from the STRUCTURE analysis were then subjected to the analysis implemented in STRUCTURE HARVESTER (Earl and vonHoldt 2012) to determine the K value based on the ∆K and the median value of lnPr(K). The online software program CLUMPAK (Kopelman et al. 2015) was used to group the estimated membership coefficients for the analysed individuals in each of the clusters.
The spatial genetic structure of all the C. pirilliformis isolates was analyzed by the haploid network analysis based on the median joining method with the default option implemented in the software package NETWORK ver.4.6.12 (www.fluxusengineering.com). The output file obtained from the calculation for transversions/transitions weights was then subjected to Post-Processing with MP calculation (Maximum Parsimony option to derive all possible shortest trees) to purge all superfluous links and median vectors, and to identify a network containing the shortest tree.
Analysis of molecular variance (AMOVA) was calculated to test the hypothesis of population differentiation between isolates from different geographic locations. A data set based on the geographic location and one on host, from which the pathogen was initially isolated, was generated and then used to compute AMOVA using R package “poppr” with 999 permutations (Kamvar et al. 2014).
Results
Fungal isolates
A Ceratocystis species with pear-shaped ascomatal bases was consistently isolated from freshly made wounds on A. mearnsii trees from ST and stumps of A. mearnsii, E. grandis and Rapanea species from GRNP in the Western Cape Province, South Africa (Table 1, 1S). Based on morphology (Barnes et al. 2003b), these isolates were tentatively identified as representing C. pirilliformis.
Phylogenetic analyses
Maximum Likelihood (ML) analyses using the combined gene regions based on part of the BT1 and TEF-1α regions resulted in a well-supported placement of the four isolates selected for confirmation of identity (CMW42018, CMW42020, CMW42236, CMW42042) with authenticated isolates of C. pirilliformis (CMW6569, CMW6579, CMW15235, CMW15236) retrieved from GenBank (Fig. 1). These results confirmed the morphological identification of the fungus as C. pirilliformis. All sequence data generated for isolates, CMW42042, CMW42336, CMW42018, CMW42020, were submitted to GenBank (KR349571 to KR349578).

RAxML phylogeny of 10 Ceratocystis species based on combined data sets of the BT1 and TEF-1α gene regions. Branch support values were indicated above and below the branches, and only bootstrap values above 50 % were presented from the analysis
Pathogenicity tests
All five isolates used in the inoculation trial produced distinct lesions on the stems of A. mearnsii trees with blue/black streaking observed in the cambium 6-weeks after the inoculations (Fig. 2). This is in contrast to the control inoculations where there were no signs of disease development (Fig. 2). There was no significant difference between lesion lengths for the five inoculated isolates, but these were all significantly different from the controls (P-value = 1.049e-09, at the 95 % confidence level) (Fig. 3). Re-isolations from the lesions resulted in C. pirilliformis isolates morphologically indistinguishable from those inoculated onto the trees.

Disease symptoms on the stems of Acacia mearnsii trees resulting from artificial inoculations with Ceratocystis pirilliformis. a: Lesion caused by C. pirilliformis, b: streaked vascular discolouration caused by C. pirilliformis, c: control (sterile malt extract agar). Arrows indicate the limits of the lesions developed after infection by C. pirilliformis

Mean lesion lengths on Acacia mearnsii trees 6 weeks after inoculation with Ceratocystis pirilliformis. The sterile agar was used in control inoculations. Standard error bars annotated with the same letters are not significantly different at P = 1.049e-09
Microsatellite amplification and scoring of alleles
Seventeen microsatellite markers were optimized (Table 3S), resulting in two multiplex panels. Of the 17 microsatellite markers, nine markers (CF17/18, CF21/22, CF23/24, CCAA80-F/CCAA81R, GACA6K-1 F/GACA6X-2R, AAG8-1 F/AAG8-1R, AAG9-1 F/AAG9-1R, CCAA9-F/CCAA9-R, CAG900-1 F/CAG900-2R) were monomorphic in all the isolates screened. The remaining eight polymorphic markers (AG7/8, CF11/12, CF13/14, CF15/16, CfCAG15F/R, DBVCAT1-1 F/WCAT1-1R, CAGDL2-5-1 F/R, CCAG15-F/R) produced between two and four alleles per marker (data not shown).
Population genetic analyses and random mating
Estimated genetic differentiation based on Rst indicated that there was no population differentiation between isolates collected from Diepwalle and Humansdrop from GRNP (equal to zero) and similarly not between Bushbuckridge, Sabie and Paulpietersburg from ESA (equal to zero) (Table 2). These isolates were, therefore, combined resulting in three larger populations (ST, GRNP and ESA) based on areas from which they had been collected (Table 1).
Gene diversity (H) values for the C. pirilliformis populations observed in this study ranged from 0.105 ± 0.058 to 0.176 ± 0.058 (Table 3). The highest level of gene diversity (H = 0.176 ± 0.058) was obtained for the ST population, followed by GRNP (H = 0.130 ± 0.051). ESA had the lowest gene diversity (H = 0.105 ± 0.058). For computed population statistics encompassing the number of different alleles (Na), the number of effective alleles (Nef) and allelic richness (AR), ST showed the highest level of population estimates (Table 3). The highest allelic richness (P), on the other hand, was obtained from ESA (P = 0.237 ± 0.106) (Table 3). A similar pattern regarding genotypic diversity was observed in which the ST population had the highest (G = 2.69, GD = 14.22, Hexp = 0.992, eMLG = 9.62), while isolates from the GRNP had the lowest level of genotypic diversity (G = 1.35, GD = 3.77, Hexp = 0.857, eMLG = 4.00) (Table 3).
The null‐hypothesis that alleles observed at different loci are randomly associated and the population is freely undergoing recombination was supported for the populations from ST and ESA with P = 0.32 and P = 0.76 respectively (Table 4). Multilocus linkage disequilibrium (I A = 1.152, r‐D = 0.288) was, however, observed in the isolates from the GRNP (P = 0.004) (Table 4).
Population structure
There was congruency from both the delta K (∆K) and the log-likelihood [Ln Pr (X|X)], yielding K = 2 as the possible number of population clusters given the data set included in this study (Fig. 4). In the CLUMPAK analysis, the single mode of clustering was obtained from K = 2 (Fig. 5). Bayesian clustering results obtained from STRUCTURE indicated that the isolates in this study could be grouped into two major clusters. All the isolates from ST and GRNP in the Western Cape region clustered into one group, whereas all the isolates from ESA (Bushbuckridge, Sabie and Paulpietersburg) made up the second cluster. These groupings clearly reflect the geographical proximity of the areas from which the isolates had been collected (Fig. 5).

Schematic representation showing the possible number of clusters based on the estimated probability of data for each K value (parameters used for the analysis: admixture model and allele frequencies correlated with three replicates for each K value), a: delta K calculated according to the method based on Evanno et al. (2005), b: graphical representation plotted based on the estimated median value and variance of probability value for each K value

Population structure of Ceratocystis pirilliformis isolates from different collection sites in South Africa. Two genetically distinct homogenous groups were defined based on the analysis implemented in STRUCTURE (K = 2). Isolates from the Western Cape (ST and GRNP) grouped together, while isolates from MP and KZN (ESA) grouped together
The haplotype network generated for the C. pirilliformis populations included in this study provided results similar to those obtained from STRUCTURE, reflecting the geographic location of the isolates (Fig. 6). Overall, two events in which closely related haplotypes (nine haplotypes from ST and five haplotypes from GRNP) were shared between ST and GRNP were observed, showing direct movement of individuals between these two geographic locations. With regards to host, isolates with the same MLH were found on A. mearnsii, E. grandis and Rapanea species, providing added support of movement of pathogen between hosts from GRNP and ESA (Fig. 6).

Haplotype network generated using the median-joining analysis based on geographic location (a) and host (b) implemented in NETWORK ver.4.6.12 for the Ceratocystis pirilliformis populations. The size of circle is proportional to the number of the haplotype used. Each colour in the circle represents the different geographical origin and host from which C. pirilliformis was initially isolated. The biggest circle consists of 15 closely related Multi-Locus Haplotypes
Hierarchical AMOVA showed that most of the variance was distributed within the population. 65 % of the variation observed was attributed to within population variation and only 35 % between populations (P-value = 0.001, Table 5).
Discussion
New collections of Ceratocystis pirilliformis arising from various hosts including a native Rapanea species and two non-native hosts (A. mearnsii and E. grandis) growing in the Western Cape Province enabled us to consider the movement of this pathogen in South Africa. Given the population diversity estimates obtained, the results suggest that the pathogen has been in the Western Cape Province for longer than in the eastern regions of the country. Along with new host records and an expanded geographic distribution for C. pirilliformis in the country, we further showed that the isolates from the Western Cape Province (ST) were able to infect A. mearnsii trees, resulting in distinct lesions on the stems of the inoculated trees. This suggests that C. pirilliformis is a putative fungal pathogen of this non-native Australian Acacia species in South Africa.
The fact that the highest level of genetic diversity was observed from the Western Cape population of C. pirilliformis raised an interesting view regarding the putative origin of the pathogen in South Africa, subsequent to its first arrival from Australia. Low levels of genetic diversity (Hallatschek and Nelson 2008), loss of alleles (Goodwin et al. 1994) and changes in reproduction modes (Goodwin et al. 1994; Taylor et al. 1999) are some of the factors that can be applied to determine whether a pathogen has been introduced into a new environment. In this regard, it can be argued that C. pirilliformis was most likely introduced to the Western Cape Province, and that it subsequently expanded its geographical range into the eastern part of South Africa given the population diversity estimates (H, Na, Nef and AR) obtained in this study.
The haplotype network analysis and Bayesian clustering results showed that isolates from ST and GRNP where A. mearnsii grows as an invasive alien tree, are closely related. These areas are also geographically closest to each other. In addition, the haplotype network analysis generated based on host clearly showed that haplotypes from A. mearnsii, Eucalyptus species and Rapanea species growing in GRNP are closely related to those from ESA. The fact that successive introductions into a new environment could arise from an invasive population that had successfully established in non-native areas is generally referred to as ‘beachhead’ effect (Lombaert et al. 2010). In this sense, some particularly virulent haplotypes of C. pirilliformis introduced to ST, possibly from Australia, might have gradually moved to GRNP, eventually leading to the expansion of host and geographical ranges with the successful establishment in ESA. This view can be supported by the fact that haplotypes shared between ST and GRNP were observed, indicating there has been direct movement of this pathogen, between these areas, most likely driven by anthropogenic activities (Santini et al. 2013; Wingfield et al. 2015).
Higher levels of genotypic diversity in comparison to relatively low levels of gene diversity observed between populations is indicative of either multiple introductions or frequent outcrossing events (Bengtsson et al. 2012; Gross et al. 2012). The impact that mode of reproduction has on population diversity is significant. In this regard, measures of genotypic diversity can be meaningful in determining the degree to which sexual reproduction occurs in a given population since the frequency of genotypes in the population is affected by mode of reproduction (Anderson et al. 1992; McDonald 1997). Relatively higher levels of genotypic diversity obtained in ESA and the fact that similar ranges of Na and Nef were observed in relation to GRNP suggest that a significant level of sexual reproduction is occurring, and this was further supported by significant linkage equilibrium based on I A and r‐D obtained from this population.
Results of this study have shown that C. pirilliformis is able to infect A. mearnsii trees, resulting in distinct lesions in the stems. The link between emerging infectious disease caused by either adapted or selected fungal genotypes with particular aggressiveness to new hosts and evolutionary mechanisms encompassing host range expansions, admixture effects and fungal introductions has been treated in numerous recent studies (Anderson et al. 2004; Slippers et al. 2005; Desprez-Loustau et al. 2007; Stukenbrock and McDonald 2008; Pariaud et al. 2009; Giraud et al. 2010; Robert et al. 2012; Barnes et al. 2014; Rius and Darling 2014; Wingfield et al. 2010, 2015). Although C. pirilliformis has never been found associated with naturally dying trees, it remains possible that it could emerge as a threat to A. mearnsii in South Africa in the future.
The significant increase in newly emerging diseases of trees is most likely attributed to large numbers of pests and pathogens being moved globally through agricultural trade and on wood packaging material especially in the forestry environment (Haack 2006; Santini et al. 2013; Wingfield et al. 2013b). The fact that haplotypes of C. pirilliformis from GRNP occur in more than one geographic location suggests that anthropogenic activities are closely linked to the movement of the pathogen in South Africa. This is especially when one considers that this pathogen is unlikely to be dispersed so widely by insects over such great distances (Heath et al. 2009; Ferreira et al. 2010). There are numerous examples of Ceratocystis species that have been accidentally introduced into new areas where they have caused significant economic losses in yield or threaten natural ecosystems (Panconesi 1999; Santini and Capretti 2000; Engelbrecht et al. 2004; Tarigan et al. 2011; Al Adawi et al. 2014). Thus, the importance of emerging new infectious disease as a consequences of fungal introductions such as the one that has been illustrated in this study should not be under-estimated.
References
Al Adawi, A. O., Al Jabri, R. M., Deadman, M. L., Barnes, I., Wingfield, B. D., & Wingfield, M. J. (2013). The mango sudden decline pathogen, Ceratocystis manginecans, is vectored by Hypocryphalus mangiferae (Coleoptera: Scolytinae) in Oman. European Journal of Plant Pathology, 135(2), 243–251.
Al Adawi, A. O., Barnes, I., Khan, I. A., Deadman, M. L., Wingfield, B. D., & Wingfield, M. J. (2014). Clonal structure of Ceratocystis manginecans populations from mango wilt disease in Oman and Pakistan. Australasian Plant Pathology, 43(4), 393–402.
Anderson, J. B., Kohn, L. M., & Leslie, J. F. (1992). Genetic mechanisms in fungal adaptation. In G. C. Carroll & D. T. Wicklow (Eds.), The fungal community: Its organization and role in the ecosystem (pp. 73–98). New York: Marcel Dekker, Inc.
Anderson, P. K., Cunningham, A. A., Patel, N. G., Morales, F. J., Epstein, P. R., & Daszak, P. (2004). Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends in Ecology & Evolution, 19(10), 535–544.
Barnes, I., Gaur, A., Burgess, T., Roux, J., Wingfield, B. D., & Wingfield, M. J. (2001). Microsatellite markers reflect intra‐specific relationships between isolates of the vascular wilt pathogen Ceratocystis fimbriata. Molecular Plant Pathology, 2(6), 319–325.
Barnes, I., Roux, J., Wingfield, B. D., O’Neill, M., & Wingfield, M. J. (2003a). Ceratocystis fimbriata infecting Eucalyptus grandis in Uruguay. Australasian Plant Pathology, 32(3), 361–366.
Barnes, I., Roux, J., Wingfield, B. D., Dudzinski, M. J., Old, K. M., & Wingfield, M. J. (2003b). Ceratocystis pirilliformis, a new species from Eucalyptus nitens in Australia. Mycologia, 95(5), 865–871.
Barnes, I., Wingfield, M. J., Carbone, I., Kirisits, T., & Wingfield, B. D. (2014). Population structure and diversity of an invasive pine needle pathogen reflects anthropogenic activity. Ecology and Evolution, 4(18), 3642–3661.
Bengtsson, S. B., Vasaitis, R., Kirisits, T., Solheim, H., & Stenlid, J. (2012). Population structure of Hymenoscyphus pseudoalbidus and its genetic relationship to Hymenoscyphus albidus. Fungal Ecology, 5(2), 147–153.
R Core Team (2014). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria, 2012. ISBN 3-900051-07-0.
de Beer, Z. W., Duong, T. A., Barnes, I., Wingfield, B. D., & Wingfield, M. J. (2014). Redefining Ceratocystis and allied genera. Studies in Mycology, 79, 187–219.
Desprez-Loustau, M. L., Robin, C., Buee, M., Courtecuisse, R., Garbaye, J., Suffert, F., et al. (2007). The fungal dimension of biological invasions. Trends in Ecology & Evolution, 22(9), 472–480.
DeVay, J. E., Lukezic, F. L., English, H., Trujillo, E. E., & Moller, W. J. (1968). Ceratocystis canker of deciduous fruit trees. Phytopathology, 58, 949–954.
Earl, D. A., & vonHoldt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4(2), 359–361.
Engelbrecht, C. J. B., Harrington, T. C., Steimel, J., & Capretti, P. (2004). Genetic variation in eastern North American and putatively introduced populations of Ceratocystis fimbriata f. platani. Molecular Ecology, 13(10), 2995–3005.
Evanno, G., Regnaut, S., & Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology, 14(8), 2611–2620.
Excoffier, L., & Lischer, H. E. (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567.
Excoffier, L., Laval, G., & Schneider, S. (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online, 1, 47–50.
Ferreira, F. A., Demuner, A. M., Demuner, N. L., & Pigato, S. (1999). Murcha de Ceratocystis em eucalipto no Brasil. Fitopatologia Brasileira, 24, 284.
Ferreira, E. M., Harrington, T. C., Thorpe, D. J., & Alfenas, A. C. (2010). Genetic diversity and interfertility among highly differentiated populations of Ceratocystis fimbriata in Brazil. Plant Pathology, 59(4), 721–735.
Fourie, A., Wingfield, M. J., Wingfield, B. D., & Barnes, I. (2015). Molecular markers delimit cryptic species in Ceratocystis sensu stricto. Mycological Progress, 14(1), 1–18.
Giraud, T., Gladieux, P., & Gavrilets, S. (2010). Linking the emergence of fungal plant diseases with ecological speciation. Trends in Ecology & Evolution, 25(7), 387–395.
Glass, N. L., & Donaldson, G. C. (1995). Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Applied and Environmental Microbiology, 61(4), 1323–1330.
Goodwin, S. B., Cohen, B. A., & Fry, W. E. (1994). Panglobal distribution of a single clonal lineage of the Irish potato famine fungus. Proceedings of the National Academy of Sciences, 91(24), 11591–11595.
Gross, A., Grünig, C. R., Queloz, V., & Holdenrieder, O. (2012). A molecular toolkit for population genetic investigations of the ash dieback pathogen Hymenoscyphus pseudoalbidus. Forest Pathology, 42(3), 252–264.
Haack, R. A. (2006). Exotic bark- and wood-boring Coleoptera in the United States: recent establishments and interceptions. Canadian Journal of Forest Research, 36(2), 269–288.
Hallatschek, O., & Nelson, D. R. (2008). Gene surfing in expanding populations. Theoretical Population Biology, 73(1), 158–170.
Halsted, B. D. (1890). Some fungous diseases of the sweet potato. The black rot. New Jersey Agriculture Experiment Station Bulletin, 76, 7–14.
Heath, R. N., Wingfield, M. J., Van Wyk, M., & Roux, J. (2009). Insect associates of Ceratocystis albifundus and patterns of association in a native savanna ecosystem in South Africa. Environmental Entomology, 38(2), 356–364.
Hurlbert, S. H. (1971). The nonconcept of species diversity: a critique and alternative parameters. Ecology, 52(4), 577–586.
Jacobs, K., Bergdahl, D. R., Wingfield, M. J., Halik, S., Seifert, K. A., Bright, D. E., & Wingfield, B. D. (2004). Leptographium wingfieldii introduced into North America and found associated with exotic Tomicus piniperda and native bark beetles. Mycological Research, 108(04), 411–418.
Kamvar, Z. N., Tabima, J. F., & Grünwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ, 2, e281.
Katoh, K., Misawa, K., Kuma, K. I., & Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research, 30(14), 3059–3066.
Kile, G. A. (1993). Plant diseases caused by species of Ceratocystis sensu stricto and Chalara. In M. J. Wingfield, K. A. Seifert, & J. F. Webber (Eds.), Ceratocystis and ophiostoma: taxonomy, ecology, and pathogenicity (pp. 173–183). St. Paul: American Phytopathological Society Press.
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A., & Mayrose, I. (2015). CLUMPAK: a program for identifying clustering modes and packaging population structure inferences across K. Molecular Ecology Resources, 15(5), 1179–1191.
Lee, D. H., Roux, J., Wingfield, B. D., & Wingfield, M. J. (2015). Variation in growth rates and aggressiveness of naturally occurring self‐fertile and self‐sterile isolates of the wilt pathogen Ceratocystis albifundus. Plant Pathology, 64(5), 1103–1109.
Lombaert, E., Guillemaud, T., Cornuet, J. M., Malausa, T., Facon, B., & Estoup, A. (2010). Bridgehead effect in the worldwide invasion of the biocontrol harlequin ladybird. PLoS One, 5(3), e9743.
McDonald, B. A. (1997). The population genetics of fungi: tools and techniques. Phytopathology, 87(4), 448–453.
Möller, E. M., Bahnweg, G., Sandermann, H., & Geiger, H. H. (1992). A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Research, 20(22), 6115–6116.
Morris, M. J., Wingfield, M. J., & Beer, C. D. (1993). Gummosis and wilt of Acacia mearnsii in South Africa caused by Ceratocystis fimbriata. Plant Pathology, 42(5), 814–817.
Nei, M. (1973). Analysis of gene diversity in subdivided populations. Proceedings of the National Academy of Sciences, 70(12), 3321–3323.
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89(3), 583–590.
Nielsen, R., Tarpy, D. R., & Reeve, H. K. (2003). Estimating effective paternity number in social insects and the effective number of alleles in a population. Molecular Ecology, 12(11), 3157–3164.
Nkuekam, G. K., Barnes, I., Wingfield, M. J., & Roux, J. (2009). Distribution and population diversity of Ceratocystis pirilliformis in South Africa. Mycologia, 101(1), 17–25.
Panconesi, A. (1999). Canker stain of plane trees: a serious danger to urban plantings in Europe. Journal of Plant Pathology, 81, 3–15.
Pariaud, B., Ravigné, V., Halkett, F., Goyeau, H., Carlier, J., & Lannou, C. (2009). Aggressiveness and its role in the adaptation of plant pathogens. Plant Pathology, 58(3), 409–424.
Peakall, R., & Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics, 28(19), 2537–2539.
Ploetz, R. C., Hulcr, J., Wingfield, M. J., & De Beer, Z. W. (2013). Destructive tree diseases associated with ambrosia and bark beetles: black swan events in tree pathology? Plant Disease, 97(7), 856–872.
Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959.
Ribeiro, I. J. A., Fumikoito, M., Paradela Filho, O., & Castro, J. L. D. (1988). Gummosis of Acacia decurrens Willd. caused by Ceratocystis fimbriata Ell. & Halst. Bragantia, 47(1), 71–74.
Rius, M., & Darling, J. A. (2014). How important is intraspecific genetic admixture to the success of colonising populations? Trends in Ecology & Evolution, 29(4), 233–242.
Robert, S., Ravigne, V., Zapater, M. F., Abadie, C., & Carlier, J. (2012). Contrasting introduction scenarios among continents in the worldwide invasion of the banana fungal pathogen Mycosphaerella fijiensis. Molecular Ecology, 21(5), 1098–1114.
Roux, J., & Wingfield, M. J. (2009). Ceratocystis species: emerging pathogens of non-native plantation Eucalyptus and Acacia species. Southern Forests: a Journal of Forest Science, 71(2), 115–120.
Roux, J., & Wingfield, M. J. (2013). Ceratocystis species on the African continent, with particular reference to C. albifundus, an African species in the C. fimbriata sensu lato species complex. In K. A. Seifert, Z. W. de Beer, & M. J. Wingfield (Eds.), The ophiostomatoid fungi: Expanding frontiers. CBS biodiversity (Vol. 12, pp. 131–138). Utrecht: CBS-KNAW Fungal Biodiversity Centre, CBS.
Roux, J., Wingfield, M. J., Bouillet, J. P., Wingfield, B. D., & Alfenas, A. C. (2000). A serious new wilt disease of Eucalyptus caused by Ceratocystis fimbriata in Central Africa. Forest Pathology, 30(3), 175–184.
Roux, J., Coutinho, T. A., Mujuni Byabashaija, D., & Wingfield, M. J. (2001). Diseases of plantation Eucalyptus in Uganda: research in action. South African Journal of Science, 97, 16–18.
Roux, J., Van Wyk, M., Hatting, H., & Wingfield, M. J. (2004). Ceratocystis species infecting stem wounds on Eucalyptus grandis in South Africa. Plant Pathology, 53(4), 414–421.
Roux, J., Heath, R. N., Labuschagne, L., Nkuekam, G. K., & Wingfield, M. J. (2007). Occurrence of the wattle wilt pathogen, Ceratocystis albifundus on native South African trees. Forest Pathology, 37(5), 292–302.
Santini, A., & Capretti, P. (2000). Analysis of the Italian population of Ceratocystis fimbriata f. sp. platani using RAPD and minisatellite markers. Plant Pathology, 49(4), 461–467.
Santini, A., Ghelardini, L., Pace, C. D., Desprez‐Loustau, M. L., Capretti, P., Chandelier, A., et al. (2013). Biogeographical patterns and determinants of invasion by forest pathogens in Europe. New Phytologist, 197(1), 238–250.
Seifert, K. A., de Beer, Z. W., & Wingfield, M. J. (2013). The ophiostomatoid fungi: Expanding frontiers. CBS biodiversity (Vol. 12). Utrecht: CBS-KNAW Fungal Biodiversity Centre, CBS.
Shannon, C. E. (2001). A mathematical theory of communication. ACM SIGMOBILE Mobile Computing and Communications Review, 5, 3–55.
Slatkin, M. (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics, 139(1), 457–462.
Slippers, B., Stenlid, J., & Wingfield, M. J. (2005). Emerging pathogens: fungal host jumps following anthropogenic introduction. Trends in Ecology & Evolution, 20(8), 420–421.
Soulioti, N., Tsopelas, P., & Woodward, S. (2015). Platypus cylindrus, a vector of Ceratocystis platani in Platanus orientalis stands in Greece. Forest Pathology, 45(5), 367–372.
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22(21), 2688–2690.
Stamatakis, A., Hoover, P., & Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Systematic Biology, 57(5), 758–771.
Steimel, J., Engelbrecht, C. J. B., & Harrington, T. C. (2004). Development and characterization of microsatellite markers for the fungus Ceratocystis fimbriata. Molecular Ecology Notes, 4(2), 215–218.
Stoddart, J. A., & Taylor, J. F. (1988). Genotypic diversity: estimation and prediction in samples. Genetics, 118(4), 705–711.
Stukenbrock, E. H., & McDonald, B. A. (2008). The origins of plant pathogens in agro-ecosystems. Annual Review of Phytopathology, 46, 75–100.
Szpiech, Z. A., Jakobsson, M., & Rosenberg, N. A. (2008). ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics, 24(21), 2498–2504.
Tarigan, M., Roux, J., Van Wyk, M., Tjahjono, B., & Wingfield, M. J. (2011). A new wilt and die-back disease of Acacia mangium associated with Ceratocystis manginecans and C. acaciivora sp. nov. in Indonesia. South African Journal of Botany, 77(2), 292–304.
Taylor, J. W., Jacobson, D. J., & Fisher, M. C. (1999). The evolution of asexual fungi: reproduction, speciation and classification. Annual Review of Phytopathology, 37(1), 197–246.
van Wyk, M., Roux, J., Barnes, I., Wingfield, B. D., & Wingfield, M. J. (2006). Molecular phylogeny of the Ceratocystis moniliformis complex and description of C. tribiliformis sp. nov. Fungal Diversity, 21, 181–201.
van Wyk, M., Roux, J., Nkuekam, G. K., Wingfield, B. D., & Wingfield, M. J. (2012). Ceratocystis eucalypticola sp. nov. from Eucalyptus in South Africa and comparison to global isolates from this tree. IMA Fungus, 3(1), 45–58.
Wingfield, M. J., de Beer, C., Visser, C., & Wingfield, B. D. (1996). A new Ceratocystis species defined using morphological and ribosomal DNA sequence comparisons. Systematic and Applied Microbiology, 19(2), 191–202.
Wingfield, M. J., Slippers, B., & Wingfield, B. D. (2010). Novel associations between pathogens, insects and tree species threaten world forests. New Zealand Journal of Forestry Science, 40, S95–S103.
Wingfield, B. D., van Wyk, M., Roos, H., & Wingfield, M. J. (2013a). Ceratocystis: emerging evidence for discrete generic boundaries. In K. A. Seifert, Z. W. de Beer, & M. J. Wingfield (Eds.), The ophiostomatoid fungi: Expanding frontiers CBS biodiversity (Vol. 12, pp. 57–74). Utrecht: CBS-KNAW Fungal Biodiversity Centre, CBS.
Wingfield, M. J., Roux, J., Wingfield, B. D., & Slippers, B. (2013b). Ceratocystis and Ophiostoma: international spread, new associations and plant health. In K. A. Seifert, Z. W. de Beer, & M. J. Wingfield (Eds.), The ophiostomatoid fungi: Expanding frontiers CBS biodiversity (Vol. 12, pp. 191–200). Utrecht: CBS-KNAW Fungal Biodiversity Centre, CBS.
Wingfield, M. J., Brockerhoff, E. G., Wingfield, B. D., & Slippers, B. (2015). Planted forest health: the need for a global strategy. Science, 349(6250), 832–836.
Acknowledgments
We thank members the Tree Protection Co-operative Program (TPCP), the National Research Foundation (NRF; Grant Specific Unique Reference Number, 78566, 83924), the THRIP initiative of the Department of Trade and Industry (DTI), and the Department of Trade and Industry (DST)/NRF Centre of Excellence in Tree Health Biotechnology, South Africa, for financial support. The Grant holders acknowledge that opinions, findings and conclusions or recommendations expressed in any publication generated by the NRF supported research are that of the author(s), and that the NRF accepts no liability whatsoever in this regard. Cultures from the Garden Route National Park were kindly made available by Mr. Alain Misse. The authors also thank the owner and viticulturist of the Knorhoek Wine Farm, James van Niekerk, who allowed us to collect samples on his farm.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lee, D.H., Roux, J., Wingfield, B.D. et al. New host range and distribution of Ceratocystis pirilliformis in South Africa. Eur J Plant Pathol 146, 483–496 (2016). https://doi.org/10.1007/s10658-016-0933-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-016-0933-7