
Abstract
Phytophthora lateralis is an oomycete responsible for Port-Orford Cedar (Chamaecyparis lawsoniana) dieback and mortality since the 1920s in western North America. It is recommended for quarantine regulation by EPPO (A2 list) and has been recently detected in Europe. In order to implement efficient control of the disease, a sensitive real-time PCR assay was developed to detect the presence of P. lateralis in plant tissues. In this study, we used the species-specific polymorphisms within the RAS-related protein gene Ypt1 to design a primer pair and a hydrolysis probe targeting the pathogen. The tool proved to be specific and inclusive, based on in silico and in vitro assessments, and could detect as few as 47 copies of target DNA. In addition, the test demonstrated its robustness and remained highly sensitive and specific under deliberately modified experimental conditions such as the hybridization temperature, or the reaction and DNA template volumes. The DNA extraction step from diseased tissues was also optimized, and the reliability of the results was ensured by a set of controls and a test targeting plant DNA that enabled the assessment of the quality of the extracts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phytophthora lateralis is the causal agent of a root rot that was first reported from Chamaecyparis lawsoniana (Port-Orford Cedar, POC) in Seattle in 1923 (Zobel et al. 1985), but was only officially described two decades later (Tucker and Milbrath 1942). The disease is now widely distributed in Western North America where it causes mortality and dieback throughout the natural range of Port-Orford Cedar, leading to severe economic and ecological losses.
In Europe, P. lateralis was first detected on C. lawsoniana in France in 1996 and 1998 (Hansen et al. 1999) and more recently from windbreak hedges in Bretagne (Robin et al. 2011). It has also been identified in Dutch nurseries (Sansford 2009) and, since 2010, in England, Scotland and Northern Ireland on diseased POC in forests, parklands and shelterbelts (Green et al. 2012). Other Chamaecyparis species and also Taxus brevifolia (DeNitto and Kliejunas 1991), Thuja occidentalis (Schlenzig et al. 2011; Green et al. 2012) and Thuja plicata (A. Schlenzig pers. comm. 2014) have also been reported as natural hosts, but with less severe symptoms. The oomycete was also discovered in 2008 in Taiwan, in soil beneath asymptomatic Chamaecyparis obtusa var. formosana trees (Brasier et al. 2010). Based on both phenotypical features and molecular data, Brasier et al. (2012) distinguished four distinct lineages within P. lateralis: two from Taiwan (J and K), one from the United Kingdom (UK) and one from North-America and Western Europe (PNW).
Zoospores of the pathogen penetrate the host through fine roots and induce collar and root lesions. Seedlings may be killed within weeks of infection and for larger trees, death may happen within a year after first appearance of crown symptoms (Winton and Hansen 2001). Aerial infections can also occur (Robin et al. 2011), due to the possible production of caducous sporangia by Phytophthora lateralis.
Although the pathogen is already present in some locations in Europe, EPPO recommends that its member countries regulate P. lateralis as a quarantine pest (http://www.eppo.int/QUARANTINE/listA2.htm). A specific and accurate detection tool is required, in order to prevent the introduction and spread of the oomycete in disease-free areas. To date, the techniques available for the detection of P. lateralis on plant samples are based either on isolation followed by morphological identification (Erwin and Ribeiro 1996) or on conventional PCR (Winton and Hansen 2001; Schena et al. 2008). However isolation and morphological identification of the pathogen is time-consuming and may lead to false negative results. Indeed, it is sometimes difficult to obtain a pure culture from a symptomatic plant sample since it requires the availability of fresh plant material and because the slow-growing mycelium of P. lateralis is easily outcompeted by faster growing organisms. The currently available conventional PCR methods alleviate these drawbacks but either lack specificity (the PCR test developed by Winton and Hansen (2001) cross-reacts with Phytophthora ramorum which may also be present on POC), or has not yet been fully adapted and validated for in planta detection (Schena et al. 2008).
The use of intronic regions of single copy genes, including the RAS-related protein gene Ypt1, proved to be effective for the differentiation of Phytophthora species (Ioos et al. 2006; Schena et al. 2008). In this respect, this region was judged to be suitable for the development of a new molecular test with enhanced performance criteria compared to the the currently available protocols.
The aim of the present work was to develop and optimize a tool for the detection of Phytophthora lateralis in plant samples, based on a highly specific real-time PCR (qPCR). A comprehensive validation process has been followed in order to assess the performance criteria of the assay according to EPPO Standard PM 7/98 (EPPO 2010). Finally, a set of ad hoc quality controls has been implemented to ensure the reliability of the results for use in of routine diagnostics.
Materials and methods
Biological material
Oomycete and fungal isolates
The isolates used in this study are listed in Table 1. P. lateralis isolates originated from several culture collections and were recovered from diseased Chamaecyparis collected in autumn 2011 on windbreak hedges in Bretagne (North-Western France). The oomycetes were isolated by plating small pieces of wood or foliage from the leading edges on PARB medium described by Robin et al. (1998) and slightly adapted (17 g Corn Meal Agar, 10 ppm Pimaricin, 10 ppm Rifampicin, 250 ppm Ampicillin, 0.015 g methyl 1- (butylamino)carbonyl-1 h-benzimidazol-2-yl carbamate, distilled water to 1 L), followed by incubation for 5 to 10 days at 22 °C in the dark. The growing hyphae were transferred to V8-agar for further morphological identification and DNA extraction. The identification was confirmed by sequencing the Internal Transcribed Spacer (ITS) using the ITS6/ITS4 primers (Cooke and Duncan 1997). True fungi were also isolated from the Chamaecyparis samples collected in the field by plating on a broad spectrum medium such as Malt agar +100 ppm Chloramphenicol. The cultures were transferred to Potato Dextrose Agar and Malt Agar for identification by morphology and ITS sequencing using ITS1/ITS4 primers (White et al. 1990).
Chamaecyparis samples
A series of 32 samples were collected in Bretagne from diseased Chamaecyparis tissues, including roots, wood, bark, branches and foliage. Each sample was chopped into small pieces (2 mm maximum) with a scalpel and the pieces were thoroughly mixed and split in two homogenous halves for: i) analysis by isolation, and ii) DNA extraction followed by molecular analysis (Table 2).
In addition, a set of 60 artificially-contaminated samples were produced in the laboratory as follows: branch pieces of Chamaecyparis lawsoniana of ca 10 mm diameter and 5 mm thickness were superficially disinfected (10 min in NaOCl 1 %), rinsed twice in sterile water and air dried; the pieces were then plated onto 10-day old V8-agar cultures of P. lateralis. After 10 days of incubation at 20 °C in the dark, the P. lateralis-infected plant material was recovered, chopped into small pieces, thoroughly mixed before distribution in 60 individual 2-mL microtubes, and stored at −30 °C until DNA extraction.
DNA extraction from pure cultures
For total DNA extraction, the aerial mycelia were scraped from the agar culture using a sterile scalpel blade and transferred into 2-mL microtubes. DNA was extracted using a DNeasy plant mini kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). The DNA concentrations were estimated using a spectrophotometer (NanoDrop 2000 – Thermo Scientific) and adjusted to 0.5 ng/μL with the AE elution buffer. The DNA extracts were kept at −30 °C until analysis. The quality of the fungal DNA extracts was assessed by conventional PCR targeting highly conserved regions: RAS primers for Phytophthora spp./Pythium spp. (RAS-E1-F/RAS-E5-R; Ioos et al. 2006) and ITS primers for other fungal species (ITS1/ITS4).
Design of primers and hydrolysis probe
Partial RAS-Ypt1 gene sequences were obtained from five P. lateralis isolates from different geographical origins by PCR using the degenerate primers RAS-E1-1F and RAS-E5-1R described by Ioos et al. (2006) and were deposited on GenBank. Two additional P. lateralis RAS-Ypt1 sequences and 13 orthologous sequences from phylogenetically close species, including P. ramorum, were retrieved from GenBank (Table 1).
All the 20 RAS-Ypt1 sequences were compared by multiple alignments, using CLUSTALW (online access: http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=/NPSA/npsa_clustalwan.html). The alignment highlighted clusters of species-specific polymorphisms and a series of forward and reverse primers and probes combinations specific for P. lateralis could be manually designed. Their melting temperature and the occurence of potential secondary structures for each oligonucleotide candidate were evaluated using Beacon Designer software (Premier Biosoft, Palo Alto, CA). Finally the qPlat-F and qPlat-R primers and the qPlat-P probe best fulfilled the technical and thermodynamical requirements for a primers/hydrolysis probe combination for real-time PCR (Bustin 2000) and were retained for further testing (Table 3).
The specificity of the qPlat-F-R-P combination was first assessed in silico by BLAST analysis on GenBank, then in vitro by qPCR with a wide range of DNA extracts from Phytophthora lateralis strains, other Phytophthora spp. or Pythium spp. isolates, and fungal species isolated from Chamaecyparis tissues (Table 1).
Implementation of quality controls
The PCR product amplified from P. lateralis genomic DNA (isolate 380-1b1) using the primer pair qPlat-F/−R (170 bp) was inserted into a pCR4-TOPO vector (Invitrogen, Carlsbad, CA) which was used to transform chemically competent TOP10 cells (Invitrogen), according to the manufacturer’s instructions. The bacterial clones were then subcultured overnight at 37 °C and the plasmids were purified using a Nucleospin Plasmid Kit (Macherey-Nagel, Düren, Germany). Based on the concatenated sequences of pCR4-TOPO and qPlat-F/-R amplicon, the molecular weight of the circularized plasmid could be determined and the number of target plasmidic copies (pc) per microliter could be calculated, according to the DNA concentration. A calibrated stock positive control solution and a tenfold serial dilution were prepared and stored at −20 °C until use.
In order to prevent cross and carry-over contaminations, preparation of the reaction mastermix and addition of the DNA extracts and positive controls were carried out in physically separated areas of the laboratory. Non-template controls were systematically included in triplicate to check the absence of contamination in all reactions.
For the rest of the experiments, the quality of the DNA extracts and the absence of inhibitory compounds were assessed by a real-time PCR using a 18S uni-F/R/P primers and probe combination that targets highly conserved regions of the 18S rDNA in plants and fungi (Ioos et al. 2009). For routine analysis, a threshold 18S Ct value (Ct18S) was experimentally determined for the 18S assay to serve as a basal reference in order to assess the quality of each DNA solution extracted from plant samples. According to Ioos and Fourrier (2011), it was considered that a DNA sample yielding a mean 18S value below or equal to Ct18S was correctly extracted and contained a sufficiently low quantity of inhibiting compounds. Preliminary 18S uni qPCR tests conducted with raw DNA extracts from Chamaecyparis samples showed that a strong inhibition effect occurred, however, a 1 in 10 dilution of the DNA extract successfully overcame the inhibition effect (data not shown). Thus all the plant DNA extracts were systematically diluted 10-fold before analysis. The Ct18S value for the routine analyses was determined by computing the mean and standard deviation for all the 18S unit Ct values obtained with 10-fold diluted DNA from 23 healthy and eight infected plant samples, as described in Ioos and Fourrier 2011; DNA was extracted following the optimized extraction protocol developed in this study.
Real-time PCR conditions
Real-time PCR reactions were performed with a Rotor-Gene 6500 (Corbett Research, Mortlake, Australia) set with an autogain optimization performed before the first fluorescence acquisition and with a LightCycler 480 (Roche, Meylan, France) for the reproducibility experiments. Amplifications were carried out in 20-μL reaction volumes using the qPCR Core kit No ROX (Eurogentec, Seraing, Belgium), consisting of molecular grade water, 1 × reaction buffer, 5 mM MgCl2, 4 × 0.2 mM dNTPs, 0.3 μM forward and reverse primer, 0.1 μM hydrolysis probe, 0.5 U of Hotgoldstar DNA polymerase, and 2 μL of template DNA. For DNA extracted from plant samples, a 10-fold dilution of the raw extract was systematically used as template.
The real-time cycling conditions for P. lateralis and 18S uni assays were identical and included an initial denaturation step at 95 °C for 10 min followed by 40 cycles of denaturation for 10 s at 95 °C and annealing-elongation for 45 s at 60 °C. The Ct values were determined using the RotorGene software version 1.7.75, setting the threshold line at 0.02. Each DNA extract was tested in triplicate and a standard deviation was calculated.
Preliminary attempts to run the qPCR test in a duplex format (qPlat-F/-R/-P and 18S uni-F/-R/-P used simultaneously) with P. lateralis target DNA diluted in a background of Chamaecyparis lawsoniana DNA resulted in an unacceptable loss of sensitivity and efficiency. The efficiency obtained for qPlat reaction in duplex with 18S uni was of 0.35 versus 0.94 in monoplex, and the reaction in duplex showed to be 10,000 times less sensitive than in monoplex (data not shown). Therefore, the qPlat-F/-R/-P and 18S uni-F/-R/-P combinations were used in separate monoplex reactions for all the experiments.
Analytical specificity and sensitivity, efficiency of the qPCR reaction
The analytical specificity was assessed with 0.5 ng μL−1 DNA extracts from 35 oomycetes and fungi isolated from Chamaecyparis lawsoniana, and from 55 isolates of P. lateralis originating from three host plants and five countries (Table 1). Furthermore, specificity was also assessed with a highly concentrated DNA extract (114 ng μL−1) from Phytophthora ramorum, which is phylogenetically the closest relative of P. lateralis. The analytical sensitivity of the qPlat test was evaluated using a tenfold dilution series of the DNA positive control, either diluted in water or in a background of C. lawsoniana DNA. A standard curve was constructed and the corresponding amplification efficiency was calculated.
Repeatability, reproducibility and robustness
Repeatability of the qPlat assay was evaluated with ten replicates of different DNA templates tested in the same run, while reproducibility was evaluated with one replicate of several DNA templates tested in ten different runs over a 4-week period, using two different thermal cyclers (Rotorgene 6500 and LightCycler 480). For each condition, intra and inter-assay coefficients of variation of Ct value were calculated. The robustness of the real-time PCR assay was assessed by measuring the effect of deliberate variations in method parameters such as DNA template volume (± 10 %), reaction volume (± 10 %) or hybridization temperature (± 2 °C and ±3 °C) on the mean Ct value yielded with target DNA (Ioos et al. 2012), and also on its specificity with non-target DNA.
Repeatability, reproducibility and robustness were assessed with plasmidic DNA controls (set at 23.6, 236 and 2360 pc/μL for repeatability and reproducibility; and 236 pc/μL for robustness) and with a 10-fold diluted DNA extract from a sample naturally infected by P. lateralis (11/379-2b). Robustness was also assessed with P. ramorum DNA adjusted to 114 ng/μL (strain LNPV390).
Comparison of plant DNA extraction procedures
Several grinding and DNA extraction options were assessed. The comparison of the DNA extraction procedures was conducted with six sets of 12 samples made of artificially-contaminated Chamaecyparis branch tissue. In an initial step, three grinding methods were tested and compared with 12 samples each. The method included: i) 2 × 1 min at 30 Hz using a 2-mL microtube with two 3-mm sterile steel beads using a beadbeater (Tissuelyser – Qiagen), ii) 2 × 1 min at a frequency of 6 units using a 2-mL microtube of Lysing matrix A (MP Biomedicals, Santa Ana, California) containing garnet matrix and ¼-inch ceramic sphere, using a FastPrep-24 homogenizer (MP Biomedicals), and iii) 2 × 1 min at a frequency of six units using a 2-mL microtube of Lysing matrix C (MP Biomedicals) containing 1-mm silica spheres, using a FastPrep-24 homogenizer. DNA extraction was performed with the DNeasy plant mini kit (Qiagen) following the manufacturer’s instructions except that the samples were ground directly with the AP1 lysing buffer. In a second step, three DNA extraction protocols were tested and compared with 12 samples each. They included the DNeasy plant mini kit (Qiagen), the Nucleospin plant II kit (Macherey-Nagel) and the PureLink Plant Total DNA purification kit (Invitrogen). For each of the three procedures, the best grinding procedure was used, considering the results of the initial step.
In order to compare the grinding and extraction procedures, the mean Ct value for the qPlat test was used as the variable. Analyses of variance and LSD-Fisher tests were performed in order to evaluate the effects of the different parameters, using XLSTAT software (version 2012.1.01; Addinsoft, Paris).
Comparative testing of the detection tools using naturally infected samples
In total, 32 Chamaecyparis lawsoniana samples showing symptoms of necrosis were collected in Bretagne (Table 2) and analyzed by three methods: i) isolation followed by classical morphological observation, ii) conventional PCR using the Ylat3F/Ylat2R PCR primers described by Schena et al. (2008) with a hybridization temperature of 58 °C, and iii) the qPlat-F/-R/-P real-time PCR. In order to prevent cross-contamination between the samples, the analytical process in the laboratory followed the forward flow requirement, and the absence of contamination during the extraction process was checked by the systematic introduction of a blank control (sterile water). Total DNA was obtained following the optimized DNA extraction protocol developed in this study (see Results section) and each DNA extract (10-fold dilution) was tested in duplicate by conventional PCR and in triplicate by real-time PCR. Some of the qPCR products were randomly picked and sequenced to verify the nature of the amplicon.
Results
Design of primers and probe, specificity, sensitivity and efficiency of the qPCR assay
Five new partial sequences of the RAS-Ypt1 gene were obtained during this study (Table 1). The DNA regions selected as targets for the primers and probe (Table 3) were 100 % conserved within Phytophthora lateralis.
The analytical specificity of the primers and probe combination was confirmed by in silico BLAST analyses with all the P. lateralis DNA sequences available on GenBank and with 13 other oomycetes DNA sequences (Table 4).
The combination of primers qPlat-F and qPlat-R with the hydrolysis probe qPlat-P was then tested in real-time PCR with a series of DNA extracts from a wide range of oomycetes, including P. lateralis, and of non-oomycetes isolated from Chamaecyparis lawsoniana. Real-time reactions using this primer-probe combination proved to be highly specific, because low Ct values were obtained for all P. lateralis isolates (Table 1), regardless of their geographical origin, whereas no Ct value was yielded with DNA from any other species. All the DNA extracts were successfully amplified by PCR using RAS-E1-F/RAS-E5-R or ITS1/ITS4 primers, thus confirming their amplifiability.
The concentrated DNA extract (114 ng/μL) from Phytophthora ramorum was also tested with qPlat-F/-R/-P and no amplification was obtained, thus supporting the high specificity of the test.
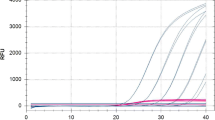
The sensitivity of the qPlat-F/-R/-P combination was measured with a 10-fold dilution series of plasmidic copies from the target in deionized DNA-free water, to yield final concentrations ranging from 2.36 to 2.36x106 pc/μL. A standard curve was constructed and the corresponding amplification efficiency was 0.99. Assays results showed a linear relationship between Ct values and log(initial concentration of the target) down to a concentration of 23.6 pc/μL, and the correlation coefficient (r2) was 0.99 (Fig. 1). The limit of detection (100 % of positive results) was 23.6 pc/μL (47.2 pc per reaction tube). The same protocol, conducted with the target DNA in a 10-fold dilution of Chamaecyparis (branch pieces) DNA extract, reached a similar limit of detection corresponding to 47.2 pc per reaction tube (r2 = 0.98). The plasmidic DNA set at the limit of detection (LOD) was then included as a control throughout the qPCR assays in order to assess the performance of the PCR runs and to ensure that the negative results were caused by an absence or a too low level of the PCR target in the DNA sample, rather than by an insufficient PCR efficiency.

Standard curve and correlation coefficient for qPlat-F/-R/-P real-time PCR assessed with dilution of target DNA in molecular-grade water. Target DNA was made of plasmids in which is inserted the P. lateralis qPlat amplicon. Final concentrations are ranging from 23.6 pc/μL to 2.36 × 106 pc/μL
Repeatability, reproducibility and robustness
For low concentrations of target DNA diluted in water as well as for a DNA from a naturally P. lateralis infected sample (11/379-2b), both the inter assay and intra-assay CVs were low with 1.78 to 2.51 % and 0.58 to 1.06 %, respectively, thus showing that the qPlat test was highly repeatable and reproducible (Table 5).
Regarding robustness, the qualitative results of the real-time PCR test were neither significantly affected by a deliberate ±10 % variation of the reaction or of the DNA template volumes, nor by a variation of the hybridization/polymerization of ±2 °C or ±3 °C (Table 6). Despite this variation, the target was detected in all cases. Even though the mean Ct value was affected by these variations of volumes or temperatures, the variations were rather limited and remained within a +/− 0.5 Ct range. The concentrated Phytophthora ramorum DNA extract was also tested in modified conditions potentially reducing stringency: decrease of the reaction volume to 18 μL, increase of the DNA template volume to 2.2 μL, or hybridization/polymerization temperature set at 57 °C and 58 °C. As a result, no amplification was obtained, thus demonstrating that the assay remained specific in spite of unfavourable reaction conditions.
Comparison of plant DNA extraction procedures
Overall, P. lateralis was successfully detected in 100 % of the DNA samples obtained, regardless of the grinding methods and the extraction kits tested, with a mean Ct value ranging from 25.7 to 32.9. Nevertheless, the Lysing matrix A with garnet matrix and ceramic sphere (FastPrep-24 homogenizer) yielded a significantly lower mean Ct value (mean Ct = 27.3, F = 48.1, p < 0.0001) than the two other grinding techniques (Table 7). When comparing the different DNA extraction kits, significant differences were also observed (F = 13.4, p < 0.0001) between Qiagen and the two other kits (Invitrogen and Macherey-Nagel) with the Qiagen kit yielding the lowest mean Ct value (Table 7). The combination Lysing matrix A and FastPrep grinding/Qiagen kit DNA extraction was therefore the best option and retained for the comparison of detection tools with field samples. Using this combination and in our conditions, the threshold Ct18S was calculated and determined at 18.8. This value should be used in the context of routine analyses to assess the quality of the plant DNA extracts.
Comparative testing of the detection tools using naturally infected samples
Out of the 32 samples tested for the presence of P. lateralis, 14 were tested negative using all three techniques. All the corresponding DNA extracts yielded a mean Ct value <21 with the 18S uni real-time PCR, which assessed the quality and amplifiability of the DNA extracts. P. lateralis was detected in 18 samples by real-time PCR, in 14 by conventional PCR (PCR), and in 11 by isolation (Table 8). All the positive cases obtained either by isolation or conventional PCR were confirmed by real-time PCR. However, for three samples, real-time PCR yielded positive results while P. lateralis could not be detected by isolation or by PCR. For these three samples, sequencing of the qPCR amplicon confirmed that the P. lateralis target DNA was amplified. In total, real-time PCR, conventional PCR and isolation yielded concordant results (positive or negative) for 71.9 % of the samples (Table 8). Real-time PCR yielded significantly more positive results than isolation (χ2 = 5.14, P = 0.023), whereas no significant difference was observed between real-time PCR and conventional PCR (χ2 = 1.33, P = 0.248), and between isolation and conventional PCR (χ2 = 0.8, P = 0.371). P. lateralis was detected by real-time PCR in tissue sampled on roots, wood, bark, branches and foliage.
Discussion
During this work, a new sensitive tool for the molecular detection of Phytophthora lateralis on plant tissues was developed and optimized to offer improved specificity and robustness. This new test does not require sub-culturing a potentially harmful organism, and manipulation of contaminated plant tissues is limited. Because this tool is DNA based, it will detect P. lateralis even if the oomycete is not in an active form which is an advantage over the isolation technique. In addition, real-time PCR provides a shorter analysis time than conventional PCR, and the tubes are kept closed after the amplification, which limits cross- and self-contamination, making it more suitable for serial analyses.
The qPlat primers-probe combination targets the intronic region of the single-copy RAS-related protein gene Ypt1, which contains sufficient interspecific but poor intraspecific polymorphism. This confirms the potential of this genomic region for the development of species-specific tools for Phytophthora, already been used by Ioos et al. (2006) and Schena et al. (2008) for conventional PCR tests. It has proven to be highly specific, since no cross-reactions were observed with 24 isolates representing 17 species of other oomycetes, including the closely related P. ramorum. In addition, the qPlat test yielded positive results with DNA from 55 P. lateralis isolates, irrespective of the origin and host plant. Unfortunately, we did not have access during this study to the isolates from Taiwan which comprise two distinct lineages (Brasier et al. 2012) and the inclusivity of the qPlat test should be confirmed by experiments in the future. Likewise, although a comprehensive evaluation of the specificity of the test has been conducted in this work, continuous monitoring would still be useful, since new species of Phytophthora are regularly discovered and described.
P. lateralis may infect different organs of the host plants, with variable severity. The method was developed for the detection of P. lateralis on symptomatic tissues, but its use for detection on asymptomatic tissues could be considered, for instance during the latent stage. This work improved the sensitivity of the test, in order to detect minute amounts of the pathogen in the plant tissue, and validated a DNA extraction protocol that yields DNA extracts with sufficient quantity and co-extracts limited amounts of inhibiting compounds. The results showed that using different DNA extractions kits and grinding procedures significantly affected the sensitivity of the test, and that a tenfold dilution of the raw DNA extract should be carried out to get an amplifiable DNA template. In order to prevent false negative results due to the presence of inhibiting compounds in the DNA extract, a 18S-uni assay was implemented in a separate real-time PCR tube. Since the PCR parameters were the same for qPlat and the 18S uni test, both assays may be conducted in the same run, making it more convenient for routine analysis.
Testing samples for the presence of a target pathogen requires very high confidence in the quality of the results, in particular in the case of a regulated pathogen. Indeed, in some occasions, the economical and ecological consequences of either a negative or a positive result may be huge. Likewise, false negative results may enable a pathogen to enter a disease-free area, whereas a false positive result may lead to the destruction of shipments or units of production. Therefore this study included a thorough validation process to evaluate the performance of the protocol within the framework of routine analysis. In line with the requirements of the EPPO Standard PM 7/98 (EPPO 2010), the test was shown to be highly repeatable and reproducible and also proved its robustness. The validation process demonstrated that slight temperature or volume variations during the reactions did not qualitatively affect the results, even when target concentrations were close to the limit of detection. This approach simulates actual conditions in laboratories in charge of routine analysis, using different equipment and with potentially different settings in micropipettes or in thermal cyclers. In all, the implementation of different quality controls such as the non template controls, the limit of detection positive control (LOD), the 18S uni qPCR test, and the experimental verification of the performance criteria make this new test fit for analysis purpose and trustworthy.
During this work, the real-time test was carried out on samples collected from diseased Chamaecyparis, in Western France. P. lateralis was successfully detected on root, wood, bark, and foliage showing symptoms. According to Hansen et al. (2000), it seems that once the pathogen is introduced and established in a forestry context, P. lateralis is virtually impossible to eradicate. It is therefore of paramount importance to prevent the introduction of this oomycete in disease free areas. Although the introduction of host plants such as Chamaecyparis are prohibited in EU (Anonymous 2000), P. lateralis might still be introduced by contaminated soil or growing media associated with non-host plants. If a sampling method and an appropriate DNA extraction protocol from soil or growing medium are developed in the future, the real-time test could also be helpful to test this kind of substrate that may harbor chlamydospores of P. lateralis. In addition, it may be postulated that the pest can be moved and introduced in disease free area by the means of symptomless plants as some chemical treatments against Phytophthora disease may suppress symptoms, while not killing the pathogen (Roth et al. 1987). Using an appropriate sampling protocol, and owing to its high sensitivity, this real-time test is expected to enable the detection of P. lateralis in such plant materials.
References
Anonymous (2000) Council directive 2000/29/EC of 8 May 2000 on protective measures against the introduction into the community of organisms harmful to plants or plant products and against their spread within the Community. O.J.L 169, 10.7.2000, 1.
Brasier, C. M., Vettraino, A. M., Chang, T. T., & Vannini, A. (2010). Phytophthora lateralis discovered in an old growth Chamaecyparis forest in Taiwan. Plant Pathology, 59, 595–603.
Brasier, C. M., Francheschini, S., Vettraino, A. M., Hansen, E. M., Green, S., Robin, C., Webber, J. F., & Vannini, A. (2012). Four phenotypically and phylogenetically distinct lineages in Phytophthora lateralis. Fungal Biology, 116(12), 1232–1249.
Bustin, S. A. (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. Journal of Molecular Endocrinolology, 25, 169–193.
Cooke, D. E. L., & Duncan, J. M. (1997). Phylogenetic analysis of Phytophthora species based on ITS1 and ITS2 sequences of the ribosomal RNA gene repeat. Mycological Research, 101, 667–677.
DeNitto, G. A., & Kliejunas, J. T. (1991). First report of Phytophthora lateralis on Pacific yew. Plant Disease, 75(9), 968.
EPPO (2010). PM 7/98 (1). specific requirements for laboratories preparing accreditation for a plant pest diagnostic activity. EPPO Bulletin, 40, 5–22.
Erwin, D. C., & Ribeiro, O. K. (1996). Phytophthora Diseases Wordwide. St Paul MN USA: American Phytopathological Society press.
Green, S., Brasier, C. M., Schlenzig, A., McCracken, A., MacAskill, G. A., Wilson, M., & Webber, J. F. (2012). The destructive invasive pathogen Phytophthora lateralis found on Chamaecyparis lawsoniana across the UK. Forest Pathology. doi:10.1111/j.1439-0329.2012.00788.x.
Hansen, E. M., Streito, J. C., & Delatour, C. (1999). First confirmation of Phytophthora lateralis in Europe. Plant Disease, 83, 587.
Hansen, E. M., Goheen, D. J., Jules, E. S., & Ullian, B. (2000). Managing Port-Orford-Cedar and the introduced pathogen Phytophthora lateralis. Plant Disease, 84, 4–14.
Ioos, R., & Fourrier, C. (2011). Validation and accreditation of a duplex real-time PCR test for reliable in planta detection of Chalara fraxinea. EPPO Bulletin, 41, 21–26.
Ioos, R., Laugustin, L., Rose, S., Schenck, N., Husson, C., & Frey, P. (2006). Usefulness of single copy genes containing introns in Phytophthora for the development of detection tools for the regulated species P. ramorum and P. fragariae. European Journal of Plant Pathology, 116, 171–176.
Ioos, R., Fourrier, C., Iancu, G., & Gordon, T. R. (2009). Sensitive detection of Fusarium circinatum in pine seed by combining an enrichment procedure with a real-time polymerase chain reaction using dual-labeled probe chemistry. Phytopathology, 99, 582–590.
Ioos, R., Fourrier, C., Wilson, V., Webb, K., Schereffer, J. L., & Tourvieille de Labrouhe, D. (2012). An optimised duplex real-time PCR tool for sensitive detection of the quarantine oomycete Plasmopara halstedii in sunflower seeds. Phytopathology, 102, 908–917.
Robin, C., Desprez-Lousteau, M. L., Capron, G., & Delatour, C. (1998). First record of Phytophthora cinnamomi on cork and holm oaks in France and evidence of pathogenicity. Annales des Sciences Forestières, 55(8), 869–883.
Robin, C., Piou, D., Feau, N., Douzon, G., Schenck, N., & Hansen, E. M. (2011). Root and aerial infections of Chamaecymaris lawsoniana by Phytophthora lateralis: a new threat for European countries. Forest Pathology, 41, 417–424.
Roth L.F., Harvey R.D., & Kliejunas J.T. (1987). Port-orford-cedar root disease. US Department of agriculture, forest service, pacific northwest region, R6-FMP-PR-294-87, 11 pp.
Sansford, C. E. (2009). Development of U.K. ⁄EU⁄EPPO pest risk analyses for Phytophthora kernoviae, P. ramorum and P. lateralis. In: Phytophthora in forests and natural ecosystems. In Proceedings of the fourth meeting of the International Union of Forest Research Organizations (IUFRO) Working Party S07.02.09: phytophthoras in forests and natural ecosystems, general technical report PSW-GTR-221. S.J. Albany, CA: U.S. department of agriculture, forest service, pacific southwest research station (pp. 139–153).
Schena, L., & Cooke, D. E. L. (2006). Assessing the potential regions of the nuclear and mitochondrial genome to develop a ‘molecular tool box’ for the detection and characterization of Phytophthora species. Journal of Microbiological Methods, 67, 70–85.
Schena, L., Duncan, J. M., & Cooke, D. E. L. (2008). Development and application of a PCR-based ‘molecular tool box’ for the identification of Phytophthora species damaging forests and natural ecosystems. Plant Pathology, 57, 64–75.
Schlenzig, A., Campbell, R., & Mulholland, V. (2011). Thuja occidentalis: a new host for Phytophthora lateralis. New Disease Reports, 24, 8.
Tucker, C. M., & Milbrath, J. A. (1942). Root rot of Chamaecyparis caused by a species of Phytophthora. Mycologia, 34, 94–101.
White, T. J., Bruns, T., Lee, S., & Taylor, J. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR protocols: a guide to method and applications (pp. 315–322). NewYork: Academic Press.
Winton, L. M., & Hansen, E. M. (2001). Molecular diagnosis of Phytophthora lateralis in trees, water and foliage baits using multiplex polymerase chain reaction. Forest Pathology, 31, 275–283.
Zobel D.B., Roth L., & Hawk G. (1985). Ecology, pathology and management of Port-Orford-cedar (Chamaecyparis lawsoniana). General Technical Report of the Pacific Northwest Forest Range Experiment Station USDA Forest Service No PNW-184, 161pp.
Acknowledgments
This research was supported financially by the French Agency for Food, Environmental and Occupational Health & Safety (ANSES).
We thank Gilbert Douzon, Dominique Sage (Pôle Santé des Forêts, Orléans, France), Maurice Nicolas, Xavier Grenie and Laurence Roche (Office National des Forêts, France) for their collaboration in the collection of the Port-Orford Cedar samples in Bretagne ; Cécile Robin (Institut National de la Recherche Agronomique, Bordeaux, France), Everett E. Hansen (Oregon University, USA), Karin Rosendahl (Nieuwe Voedsel en Waren Autoriteit, Wageningen, the Netherlands) and Alexandra Schlenzig (Science and Advice for Scottish Agriculture, Edinburgh, Scotland) for supplying numerous P. lateralis isolates used in this study; Claude Husson (INRA Champenoux, France) for providing healthy C. lawsoniana material.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Schenck, N., Fourrier-Jeandel, C. & Ioos, R. A robust and specific real-time PCR tool for the detection of Phytophthora lateralis in plant tissues. Eur J Plant Pathol 146, 231–244 (2016). https://doi.org/10.1007/s10658-016-0909-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-016-0909-7