
Abstract
The strong association between Phytophthora cinnamomi and the mortality and decline of Quercus suber and Q. ilex subsp. rotundifolia has been known for two decades. The ability of elicitins secreted by this pathogen to trigger defence responses in these Quercus against itself was evaluated in this work. Biomass quantification by quantitative real-time PCR revealed a significant decrease in pathogen colonization of Q. suber roots after 24 h pre-treatment with α- and β-cinnamomin. In Q. suber and Q. ilex roots pre-treated with α-cinnamomin, hyphae were unable to reach and colonize the vascular cylinder and showed cytoplasmic disorganization in all the roots observed as contrasted with non-pre-treated roots. The pathogen was restricted to the intercellular spaces of the cortical parenchyma and the concomitant accumulation of electron dense materials was observed in contact with the hyphae. Furthermore, ROS (reactive oxygen species) production and the enzymatic activities of superoxide dismutase, catalase and peroxidase were compared in infected and non-infected Quercus roots in time course trials. There was a significant increase in the production of hydrogen peroxide (H2O2) and superoxide anion (O2 •-) and an enhanced activity of the enzymes in infected roots was observed at each time point. When comparing with elicitin non-treated roots, the α-cinnamomin-treated roots in interaction with P. cinnamomi showed a decrease in ROS accumulation and an increase of the enzyme activities. The overall results were consistent with an induction by the cinnamomins which initiated defence responses against the pathogen invasion of roots. Finally, elicitins were immunolocalized in the contact zone of P. cinnamomi hyphae with epidermal host cells, plasmalemma outer cytoplasm and around the intracellular hyphae in the vacuoles of invaded epidermal cells.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The destructive power of Phytophthora cinnamomi has been abundantly documented. This phytopathogen whose action covers a wide host range and reaches large regions of the globe has been responsible for massive destruction of the Jarrah forest (Eucalyptus marginata) in southwestern Australia (Shearer and Tippett 1989), and the devastation of thousands of chestnut trees in the southeastern United States (Castanea dentata), in northern Portugal and in Galicia (Spain) (C. sativa) (Crandall 1950). In western Mediterranean regions, in particular, P. cinnamomi has been the main biotic agent associated with the severe decline of evergreen oaks such as cork oak (Quercus suber L.) and holm oak (Quercus ilex subsp. rotundifolia L.) (Brasier et al. 1993; Sánchez et al. 2002; Caetano 2007). This decline endangers the future of cork production, which is entirely provided by Mediterranean countries with predictable and disastrous economic, social and environmental consequences. The pathogenicity of P. cinnamomi in Mediterranean and temperate oaks has been documented (Robin et al. 1998). The pathogen appears to play an important role in the necrotic activity observed on feeder roots, leading to the reduction of the number of roots available for water and nutrient uptake. More recently it has been reported in cork oak that P. cinnamomi hyphae spread inter- and intracellularly throughout the cortical parenchyma and invaded the vascular cylinder. Hyphae also filled the xylem and phloem vessels, leading to their occlusion (Medeira et al. 2012a; Horta et al. 2010).
Oomycete species take the control of molecular processes in their host plants by involving virulence or avirulence molecules, known as effectors. For example, most Phytophthora and a few Pythium species specifically secrete large amounts of elicitins, a group of unique highly conserved proteins whose molecular roles have been investigated and constitute a matter of debate (reviewed by Ponchet et al. 1999; Oßwald et al. 2014). Elicitins are able to facilitate infection (virulence factors), appear to be required for the colonization process, and are determinants for the aggressiveness of the pathogen. For instance, it was recently reported for the Q. suber-P. cinnamomi pathosystem using a β-cinnamomin (β-CIN) knock-down P. cinnamomi strain (Horta et al. 2010) and shown for the role of citricolin from P. plurivora in the infection of beech roots (Oßwald et al. 2014), and also suggested by the positive correlation of virulence with ram-a2 expression in P. ramorum (Manter et al. 2010). Elicitins were shown to induce defence responses in plants (avirulence factors or elicitors) (Bonnet et al. 1996). The first studies by Ricci et al. (1989) showed that cryptogein and capsicein cause a hypersensitive response (HR) in tobacco as well as systemic acquired resistance (SAR) to normally virulent pathogens. Later, it was proposed that with the exception of those belonging to the families Solanaceae and Cruciferae, most plant species lack the capacity to respond to elicitins (Grant et al. 1996). However, oligandrin, an elicitin-like protein secreted by Pythium oligandrum, was shown to induce systemic resistance to Fusarium oxysporum and Botrytis cinerea in tomato and grapevine, respectively (Benhamou et al. 2001; Mohamed et al. 2007). Also, recent histological and ultra-structural studies (Medeira et al. 2012a, 2012b), assessing of changes in fatty acid composition of roots and leaves, and gas exchanges and fluorescence analysis (Medeira et al. 2012a) have documented that cryptogein and capsicein can trigger cellular responses in cork oak roots (Medeira et al. 2012a) and α-cinnamomin (α-CIN) in Castanea sativa roots (Medeira et al. 2012b) during P. cinnamomi infection. This strongly suggests that a resistance defence arose against the pathogen upon elicitin treatment.
The biological role of sterol carrier activity has also been assigned to elicitins due to their peculiar structural features consisting of a hydrophobic cavity that may accommodate a sterol molecule (Mikes et al. 1997; Rodrigues et al. 2006). Sterol loading from the plasmalemma allows the binding of the elicitins to their receptor, which then activates defence mechanisms in plants (Blein et al. 2002).
Previous studies showed that plants produce harmful reactive oxygen species (ROS) under pathogen attack, including the production of superoxide anion (O2 ⋅−) and hydrogen peroxide (H2O2), both produced and released at the site of pathogen ingress (Apel and Hirt 2004). High concentrations of ROS can result in oxidative damages, whereas low level of ROS production can serve as signaling molecules and induce antimicrobial activity and defence mechanism in plants (Lamb and Dixon 1997). In order to avoid oxidative injury in cells, plants contain scavenging enzymes that modulate the ROS production. Various enzymes systems participate in ROS metabolism during pathogen attack in plants. Major ROS scavenging enzymes such as superoxide dismutase (SOD) and catalase (CAT) are produced to avoid cellular disintegration caused by ROS (Mittler 2002). Furthermore, it is well established that peroxidases (POD) are involved in defence processes against plant pathogens catalysing the formation of lignin and other oxidative phenols that have been associated with the formation of defensive barriers against the pathogens (Vance et al. 1980).
In this study, we applied three approaches to evaluate the effect of cinnamomins, produced by Phytophthora cinnamomi, on the infection process of evergreen Mediterranean oak roots by this same pathogen: 1) cytological characterization of elicitin induced protection in Q suber and Q. ilex subsp. rotundifolia, 2) pathogen biomass quantification, and 3) evaluation of ROS formation and of antioxidant enzyme reactions during initial responses of Q. suber against the invading pathogen P. cinnamomi, after α-CIN or β-CIN treatment. In addition, the cinnamomins were localized in Q. suber roots by immunocytochemical techniques.
Materials and methods
Plant material, elicitin treatments and inoculation with P. cinnamomi
P. cinnamomi isolate XX-1B was isolated from the rhizosphere of cork oak trees in declining stands in southern Portugal and was grown on Difco potato-dextrose agar (PDA, Difco Laboratories, Detroit, MI, USA) in 9-cm Petri dishes for 3–5 days, in dark conditions, at 25 °C.
The cytological studies, used cork oak and holm oak seedlings germinated from acorns collected in Baixo Alentejo, Portugal, grown in vermiculite in a greenhouse at 22 °C with a 14 h light period, and 60-80 % relative humidity, for 2 months. Ten plants of each species were removed from the substrate and their primary and lateral fine roots were washed and immersed in an aqueous solution of α-CIN (0.5, or 10 μM) for 24 h. After α-CIN treatment, five plants were inoculated with P. cinnamomi by placing discs (1 cm diameter) of a young culture in contact with the root system. The other five plants were treated similarly but with P. cinnamomi-free potato dextrose agar (PDA) discs. Two control groups of cinnamomin non-treated plants were prepared the following way: 1) infecting five plants in absence of cinnamomin, and 2) taking five additional plants cinnamomin non-treated and non-infected. Root samples were collected 2 days after pathogen inoculation. All the samples were collected at the same time to be processed later for microscopy observations.
For the quantification of the pathogen colonization, seeds of cork oak were germinated (20 in total) in vermiculite until 2-week-old roots reached approximately 10 cm. The roots were removed from the substrate and submitted to five different conditions for 24 h: immersion in aqueous solutions α-CIN and β-CIN at two different concentrations: 0.5 μM (5 μg ml−1) and 10 μM (100 μg ml−1) of each elicitin and in water. Four plants per condition were used. The roots were inoculated by placing a 2 mm diameter plug of 5-day-old mycelia of P. cinnamomi, grown on 20 % V8-juice agar, at the tips of the roots used, then covering the roots with filter paper pre-moistened with sterile distilled water for 24 h.
The reactive oxygen burst study used α-CIN-treated (0.5 μM) and non-treated groups that were prepared similarly to the method described above. Two-month-old cork oak seedlings were used and the roots were collected at different time points (6, 12, 18, 24, 36 and 48 h) after infection with P. cinnamomi (discs of 5-day-old mycelia, 1 cm in diameter) and used for enzymatic assays. Three to five replicates were used at each time point and the overall experiment was repeated three times.
α and β-cinnamomins
Expression and purification of these cinnamomins was as described in Rodrigues et al. (2006) and Archer et al. (2000). Briefly, the ORF of α-CIN or β-CIN was amplified by PCR from the recombinant vector pTopo α-cin or β-cin obtained by cloning the α-cin or β-cin gene in the PCR II®-TOPO vector (Invitrogen). The PCR product was inserted into the methylotrophic yeast Pichia pastoris secretion vector pIC9K downstream to and in frame with, the DNA sequence encoding the α-mating factor pre-pro secretion signal sequence of Saccharomyces cerevisiae.
In the case of β-CIN, P. pastoris cells were harvested in a tubular centrifuge at 30,000 x g and the supernatant, diluted with Milli RX water until a conductivity of 6 mS, was loaded onto an anion exchange column filled with 300 ml of Streamline Q XL (Pharmacia) equilibrated with 20 mM NaH2PO4 at pH 5.0. The flow through was then adjusted to pH 3.5 and loaded into a cation exchange column (Fractogel SO3 −, Merck KgaA xk26/60), which was equilibrated with 20 mM NaH2PO4 at pH 3.5 The column was eluted with 500 mM NaCl in the same buffer, using a linear gradient in 5 column volumes. The β-CIN protein was eluted between 100 and 150 mM NaCl.
Further purification was achieved using a endotoxin removal column (FRACTOGEL DEAE, Merk KgaA) equilibrated with 20 mM NaH2PO4 pH 3.5.
In the case of α-CIN the same procedure was applied with following the differences: the supernatant was concentrated 100 times using an ultrafiltation membrane system (cut-off 8 kDa) with tangential flow in two successive steps and an YM-10 membrane (cut-off 10 kDa) under a pressure of 3 bars and instead of a cation column, an anion column was used (Q Sepharose fast flow- Pharmacia- column equilibrated in 10 mM sodium acetate, (pH 5.7) 1 M NaCl buffer. Product identification and purity assessment were done by Western Blot and SDS PAGE. The yield ranged from 174 mg (α-CIN) to 47 mg (β-CIN) of purified protein per liter of culture.
Tissue processing for microscopy
Well washed Q. suber or Q. ilex subsp. rotundifolia fine roots (4 per seedling selected randomly) were cut into fragments of about 1 mm3 in the inoculated zone or above the α-CIN immersed zone (in cinnamomin-treated samples) or in the corresponding root level in the control samples. The root fragments were fixed in 3 % glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.3, overnight at 4 °C. Then, the samples were thoroughly washed for 30 min in the same buffer, post-fixed with 1 % osmium tetroxide in 0.1 M sodium cacodylate buffer for 1 h, dehydrated in an ethanol gradient to absolute ethanol and embedded in Spurr’s resin. Alternatively, for light microscopy (LM) the root fragments were fixed in 0.1 M cacodylate buffer (pH 7.2) containing 3 % (vol/vol), glutaraldehyde and 4 % (wt/vol) paraformaldehyde, for 4 h at 4 °C. Fixed tissues were washed in the same buffer, dehydrated in an ethanol gradient to absolute ethanol and embedded in LR White resin (London Resin Company). Thick sections (1–2 μm) and thin sections (60–80 nm) were cut in a microtome (Leica Ultracut, Austria), using diamond knives. Thick sections were either stained with 0.05 % toluidine blue in 1 % sodium carbonate and observed by LM (Leica DM LD microscope, Austria) or unstained and observed by phase contrast microscopy the same microscopy system. Thin sections were stained with an aqueous saturated solution of uranyl acetate for 45 min and post-stained in a 3 % (w/v) aqueous lead citrate solution at room temperature for 30 min. The observations were made with a transmission electron microscope (TEM; FEI Morgagni 268D, USA) at 80 kV. In total, per treatment, 400 thick sections and 100 ultrathin sections were examined under LM and TEM, respectively.
Immunocytochemical localization of α-cinnamomin
For the localization of cinnamomin in Q. suber roots in interaction with P. cinnamomi, the roots were inoculated by placing a 2 cm2 V8 agar plug containing actively growing mycelium in contact with the root surface for 2 days. Root fragments were collected from the inoculated zone and fixed in 0.1 M cacodylate buffer (pH 7.2) containing 0.5 % (v/v), glutaraldehyde and 4 % (w/v) paraformaldehyde, for 2 h at 4 °C. Fixed tissues were washed in the same buffer and treated with 50 nM ammonium chloride in 10 nM cacodylate buffer for 30 min, in order to hinder the non-specific attachment of antibodies to free aldehyde groups. After dehydration in a graded series of ethanol, tissues were progressively infiltrated in LR White medium grade (London Resin Company) and were polymerized for 24 h at 50 °C.
Thin sections from pieces of roots embedded in LR White were collected on formvar-coated nickel grids and then successively incubated for 30 min in 10 mM Tris–HCl buffer, pH 7.4 with 0.9 % (w/v) NaCl (TBS buffer) containing 1 % (w/v) dried milk and 10 % (v/v) normal goat serum and finally incubated for 1 h at 37 °C with the polyclonal antibody to cinnamomin (supplied by Laboratoire d’Hormonologie, C.E.R.-Marloie, Belgium). After thoroughly washing in the same buffer, the grids were incubated for 1 h at room temperature with a goat anti-mouse immunoglobulin G labeled with 10 nm colloidal gold particles (Sigma) washed in pure sterilized water and counterstained with 3 % (w/v) aqueous uranyl acetate for 30 min.
The labeling specificity was assessed through the following control experiments: 1) omission of the primary antibody incubation step, 2) incubation with the antiserum previously adsorbed with the corresponding antigen for 3 h at room temperature.
For the localization of α-CIN absorbed by Q. suber roots upon 24 h of immersion in a solution 10 μM α-CIN in water, fragments of about 1 cm3 were excised from the roots, 1 to 2 cm above the application point of the elicitin and were fixed, embedded, and processed as described for immunocytochemical localization of the α-CIN secreted by P. cinnamomi in the interaction with the host.
Quantification of Phytophthora cinnamomi biomass in Quercus suber roots
Plasmid DNA
Plasmid DNA containing the Myc mouse gene, was prepared using mouse. Myc primers (MYC Ex2-FOR 5′-GCTGGAGATGATGACCGAGT-3′) and (MYC-Ex2-REV 5′-AACCGCTCCACATACAGTCC-3′) were designed and used for the amplification of a fragment 119 bp long. PCR reactions (50 μl) were performed using 20 ng of gDNA, 5 μl 10× Dream Taq Buffer which includes 20 mM MgCl2, 5 μl dNTPs (dATP, dCTP, dGTP and dTTP, 2 mM each), 10 mM of primers and 1.25 U of DreamTaqTM DNA polymerase (Fermentas). All reactions were carried out on a MyCycler thermocycler (BioRad), using the following conditions: initial denaturation at 94 °C for 5 min, 30 cycles at 94 °C for 30 s, annealing step at 60 °C for 30 s and 72 °C for 1 min, followed by a final extension at 72 °C for 10 min. PCR products were purified with the Qiaquick PCR Purification Kit (Qiagen) and cloned into the pCRII Topo vector, with the TA Cloning Kit (Invitrogen). Plasmid DNA was extracted using the PureLink™ Quick Plasmid Miniprep Kit (Invitrogen). Manufacturer’s instructions for these kits were followed throughout.
The plasmid DNA was sequenced in an Applied Biosystems 3130 × l Genetic Analyzer carrying out reactions with BigDyeTerminator Kit from Applied Biosystems.
Plant DNA extraction using plasmid DNA as an internal control
Genomic DNA was extracted from α-CIN and β-CIN-treated and non-treated roots, 24 h after inoculation (hpi), using a modified cetyltrimethylammonium bromide-based method. Powdered root tissues (50 mg) were homogenized in CTAB buffer [2 % (w/v) CTAB; 25 mM EDTA, pH 8.0; 100 mM Tris–HCl, pH 8.0; 0.05 % spermidine; 2 % polyvinylpolypyrrolidone; 2 M NaCl] containing 3 % β-mercaptoethanol and incubated at 65 °C for 30 min. After centrifugation at 10,000 rpm for 10–20 min, 2 μl of RNase A (10 mg ml−1) was added to the collected upper phase and tubes incubated at room temperature for 30 min. In this step 1 ng of Myc plasmid DNA (containing myelocytomatosis oncogene − Myc) was added to all samples. One volume of chloroform:isoamylalcohol (24:1) was added to the supernatant. After centrifugation at 10,000 rpm for 5 min this step was repeated. Then, the volume of supernatant was determined and one volume of ice-cold isopropanol was added and mixed, and stored at −20 °C for 10 min, then centrifuged at 12,000 x g for 2–5 min at −4 °C. After centrifugation and rinsing with 70 % ice-cold ethanol, the extracted DNA pellets were dissolved in 50–100 μl sterile DNasefree water. A Nanodrop ND-1000 spectrophotometer (Thermo Scientific) was used to measure the quantity and assess the purity of genomic DNA.
Primer design
Phytophthora cinnamomi primers (forward 5′-GCTAGCAAGCACGTATGAGG-3′ and reverse 5′-CGCCCCAACTATACGACAAC-3′) were employed for the amplification of a PDN gene (FJ493007) fragment with an amplicon size of 90 bp (Eshraghi et al. 2011). PCR reactions were carried out using 20 ng of gDNA as template in a total volume of 50 μl containing 5 μl 10× Dream Taq Buffer, 20 mM MgCl2, 5 μl dNTPs (dATP, dCTP, dGTP and dTTP, 2 mM each), 10 mM of primers and 1.25 U of DreamTaqTM DNA polymerase (Fermentas). Amplification reactions were carried out on a MyCycler thermocycler (Biorad), programmed for 5 min at 94 °C; 30 cycles at 94 °C for 1 min, 30 s at 60 °C, 1 min at 72 °C; and a final extension of 10 min at 72 °C. The specificity of the plasmid and P. cinnamomi primers was assessed by comparing the sequence of the primers with the known genome sequences of Q. suber, P. cinnamomi and Myc gene and running PCR on all these DNAs as templates (genomic DNAs of P. cinnamomi and of Q. suber, and plasmid DNA). On the basis of these analyses the primers were judged to be specific for their target DNA templates.
QPCR amplification
Quantitative RT-PCR reactions were conducted in an iQ5 multicolour Real-Time PCR Detection System (Bio-Rad, USA). All DNA samples were analyzed using SsoFast™ Evagreen® supermix kit (Biorad). For each sample, 20 ng of DNA template was mixed with 7.5 μl of SsoFastTM Evagreen® supermix, 1 μl of forward primer and 1 μl of reverse primer (final concentration 50 nM each primer) in a final volume of 15 μl. The following standard thermal cycling profile was used: 95 °C for 10 min; 30 s at 95 °C, 40 cycles of 1 min at 60 °C annealing temperature and 30 s at 72 °C. To confirm that the signals were the result of a single amplified product, melting curve analysis was performed after the run by increasing the temperature of 0.5 °C by cycle from 65 to 95 °C.
The amplification efficiency of the target gene was checked by the slope of a standard curve constructed from amplification of 10-fold dilution series (ranging from 10,000 to 1 pg) of DNA extracted from P. cinnamomi and 10-fold dilution series (ranging from 1,000 to 0.1 pg) of plasmid DNA.
Colonization was expressed as the ratio between pathogen PDN and plasmid DNA copy number. The ratio was based on the mean value of four biological replicates.
Measurement of reactive oxygen species and of antioxidant enzymes
Hydrogen peroxide
The hydrogen peroxide content was determined in Q. suber roots during the time course of P. cinnamomi contact up to 48 h, according to Cakmak and Marschner (1992). The assay was modified using peroxidase as a catalyst and guaiacol as a substrate. Fresh root tissue (0.5 g) was immersed for 10 min in 10 ml deionised water. The reagent mixture used to quantify hydrogen peroxide contained 0.05 % (v/v) guaiacol and horseradish-peroxidase (2500 U ml−1) dissolved in 25 mM sodium phosphate buffer (pH 7.0). H2O2 in the medium was measured by incubating 1 ml of the root suspension in water with the same volume reagent mixture for 15 min at room temperature in the dark. The reaction turns the reagent mixture to an unstable brown color and absorbance was measured immediately at 450 nm. Commercial hydrogen peroxide (37 % pure, Merck, Germany) was used to plot a standard calibration curve using solutions with known concentrations. Control samples were assayed in the same way as the inoculated samples.
Superoxide anion radical
The production of the radical superoxide anion (O2 •−) was measured in Q. suber roots for up to 48 h during the time course of P. cinnamomi contact using an assay based on reduction of the nitro blue tetrazolium dye sodium, 3′-(1-[phenylamino-carbonyl]-3,4-tetrazolium)-bis(4-methoxy-6-nitro) benzene-sulfonic acid hydrate (XTT). Fresh root tissue (0.5 g) was homogenized with 1.0 ml of 50 mM Tris–HCl buffer (pH 7.5) and centrifuged at 14000 × g for 5 min. XTT was added at a final concentration of 0.5 mM to 0.5 g root tissue. The samples were inoculated for 10 min and 1 ml of supernatant was collected for analysis of XTT formazan production at 470 nm on a spectrophotometer (Pharmaspec uv-1700, Shimadzu, Kyoto, Japan) (Able et al. 1998).
Extraction and quantification of proteins
Approximately 1.0 g fresh weight (FW) of ground root tissue was homogenized in 4 ml of 80 mM phosphate buffer (pH 7.0), 1 mM benzamidine, 0.1 % (v/v) 2-mercaptoethanol, 1 mM EDTA, 0.1 % (v/v) Triton X-100, and 1 % (w/v) polyvinylpolypyrrolidone at 4 °C. The homogenates were centrifuged at 14,000 × g for 20 min at 4 °C, and the supernatants were recovered. All enzymatic assays were performed using freshly prepared protein extracts that were kept on ice until analysis. Proteins were quantified by using a Micro BCA Protein Assay Kit (Thermo Scientific, USA). Micro BCA Working Reagent (WR) was prepared by mixing 25 parts Micro BCA Reagent MA, 24 parts Micro BCA Reagent MB, and one part Reagent MC. The unknown samples (diluted 1/100 and 1/1000) were added at 1 ml volume to 1 ml of WR and mixed thoroughly and incubated at 60 °C in a water bath for 1 hour. The samples were cooled to room temperature and the absorbance was measured at 562 nm on a Perkin Elmer spectrophotometer (MBA 2000, Waltham, MA, USA). A set of diluted Albumin (BSA) standards were prepared (from 1 to 40 μg ml−1) to construct a standard calibration curve.
Enzymatic assays
Total peroxidase
Guaiacol was used as substrate for the analysis of total peroxidase activity (POD). The reaction mixture (1 ml) consisted of 990 μl guaiacol solution containing 0.25 % guaiacol (v/v) in 10 mM sodium phosphate buffer (pH 6.0) and 0.125 % H2O2 (v/v); 10 μl of extracted protein was added to this mixture. POD activity was observed as an increase in absorbance (Abs) at 470 nm. The reaction was followed for 1 min. The linear ∆Abs/min/mg protein was used as a measure of relative POD activity.
Superoxide dismutase (SOD)
The SOD (EC 1.15.1.1) activity was determined as described by Beyer and Fridovich (1987). This method is based on the inhibition of SOD activity in the reduction of nitroblue tetrazolium chloride (NBT) by the superoxide anion radicals generated photochemically. The reaction mixture consisted of 100 mM phosphate buffer (pH 7.8), 0.2 mM EDTA, 19.8 mM L-methionine, 0.05 % (v/v) Triton X-100, 57 μM NBT, 0.9 μM riboflavin, and 50–250 μl protein extract. After 6 min of incubation at 30 °C under continuous light, absorbance was read at 560 nm. One unit (U) of SOD activity is defined as the amount of enzyme required to cause 50 % inhibition of NBT reduction under the assay conditions as described.
In order to determine the % inhibition for the test sample(s) the following formula was applied:
Catalase (CAT)
Catalase (CAT, EC 1.11.1.6) activity was assayed following a method by Aebi (1984) based on the decomposition of H2O2. Reactions were performed in 80 mM phosphate buffer (pH 7.0) containing 1 mM H2O2 and initiated by adding 20–50 μl of protein extract. The decrease in absorbance was evaluated at 240 nm and 25 °C. One unit (U) of CAT activity is defined as the amount of enzyme necessary to decompose 1 μmol min−1 H2O2 under the above assay conditions [ε (H2O2; 240 nm) = 39.43 M−1 cm−1].
Statistical analysis
Data was analyzed using SPSS (SPSS ver. 20). One-way analysis of variance (ANOVA) with Tukey’s post hoc tests was used to reveal significant differences between infected and control plants for each treatment in biomass quantification and ROS production studies. In addition, data from ROS production was further analyzed to compare cinnamomin-treated and non-treated samples at the indicated time points, using t-test implemented in SPSS.
Results
Histological and cytological studies
Cork oak and holm oak roots infected with Phytophthora cinnamomi
Cyto-histological studies of the 2-month old seedlings roots infected with mycelia were done at 48 hpi. The pattern of root cells colonization was very similar for the two oak species. Forty eight hpi of 2-month-old seedling roots with mycelia, the pathogen had penetrated the epidermal cells and spread inter- and intracellularly throughout the cortical parenchyma (Fig. 1a). Intracellular hyphae were frequently observed expanding in large aggregates. The disruption of host cell walls occurred and swelled hyphae were released into the intercellular spaces of the cortical parenchyma. In the vascular cylinder the pathogen colonized and damaged the vascular tissues leading to the disruption (Fig. 1b) and to the occlusion of xylem vessels (Fig. 1c).

(LM) Cork oak and holm oak roots infected with P. cinnamomi. a (holm oak): penetration of epidermal cells by the pathogen and its spreading through the cortical parenchyma; the pathogen expanded inside the cortical cells and formed aggregates (*) of swelled hyphae in the vascular cylinder. b host cell wall disruption (arrow) and degradation of extensive areas in holm oak. c (cork oak): the invasion of the pathogen led to xylem occlusion. P pathogen; VC vascular cylinder
Ultrastructural observations showed the active metabolic aspect of the invading pathogen with the dense cytoplasm, rich in ribosomes and mitochondria with tubular-vesicular cristae, endoplasmic reticulum, lipid bodies and vacuoles and the severely damaged root tissues with host cell wall and cytoplasm degradation or necrosis visible around zones in contact with the hyphal walls (Fig. 2).

(TEM) Cork oak and holm oak roots infected with Phytophthora cinnamomi. The pathogen inside cortical cells exhibited high metabolic activity expressed by large number of organelles. a (cork oak), b and c (holm oak): lipid bodies (LB), mitochondria (M), vacuoles (V) and endoplasmic reticulum (ER). In c a restriction of hyphal diameter at the point of pathogen (P) penetration in a collapsed host cell (HC). An extra-hyphal matrix (arrow) formed by fibrillar electron-dense material surrounds the hyphae
Constrictions of hyphal diameter were observed at the sites of pathogen penetration into the host cells, but wall appositions were not seen. An extra-hyphal matrix formed by fibrillar electron-dense material that envelopes the hyphae, was seen frequently in some hyphal penetrations, as well as the collapse of the host cells (Fig. 2c).
Cinnamomin root treatment
Cork and holm oak roots treated with α-CIN for 24 h did not reveal any signs of injury, but an osmiophilic increase in vacuoles, compared with the control, was observed. The ultrastructure of the parenchymal cells was well preserved and did not show any signs of damage of plasmalemma, tonoplast or organelle membranes (Fig. 3).

Cork oak roots treated with α-CIN. a (LM) Part of a root section in which no visible signs of cellular damage was observed. An osmiophylic increase of the vacuoles (*) is visible in comparison with control plants (in c). Cortical cells (C), endodermis (E) and vascular cylinder (VC) are also visible. b and d (TEM) Well preserved ultrastructure of the internal cortical parenchymal cells, mitochondria (M), plasmalemma (PL), tonoplast (T) in cinnamomin-treated roots. Accumulation of electron-dense fibrillo-granular materials (*) in the intercellular spaces of the cortex is visible in d, not observed in the controls. e (TEM) control non-cinnamomin-treated root
Infection of cinnamomin treated roots
Responses of cork and holm oak α-CIN pre-treated roots to the infection by P. cinnamomi are very similar. Within 48 h of inoculation the pathogen penetrated the epidermal cells progressing mainly through the intercellular spaces of the cortical parenchyma. The hyphae showed signs of cytoplasmic disorganization and, in all observed roots, were unable to reach and colonize the vascular cylinder (Fig. 4).

(LM) Cork oak and holm oak roots treated with α-CIN and infected with Phytophthora cinnamomi (P). a (holm oak) Penetration of the epidermal cells by the pathogen. b (holm oak) Progression of the pathogen through the cortical parenchyma, mainly intercellularly. c (holm oak) The hyphae did not reach the vascular cylinder (VC). (CP = Cortical parenchyma). d (cork oak) green autofluorescence observed under UV. Localization of the infection shown by circles. e (cork oak) Autofluorescent materials (arrows) localized around the pathogen (P) and along host cell walls. f (cork oak) Accumulation of vacuolar autofluorescent materials (arrow). g (cork oak) When cortical cells were stained with toluidine blue the vacuolar precipitates appeared bright green (arrows). The pathogen (P) showed signs of disorganization close to the cells containing the precipitates
The restriction of the pathogen invasion to the cortex was observed in association with auto-fluorescent molecules under UV light (Fig. 4d to f). These deposits were found around the hyphae in the intercellular spaces or inside the parenchymal cells, by auto-fluorescence blue green in the vacuoles and walls of the invaded cortical cells or in the cells near the hyphae (Fig. 4f). When these cells were stained with toluidine blue the vacuolar precipitates appeared bright green (Fig. 4g).
Ultrastructural observation of the hyphae in the intercellular spaces showed evidence of pathogen degradation including the loss of membrane and organelles integrity (plasmalemma, tonoplast degradation) and loss of ribosomes. Osmiophylic fibrillo-granular materials were observed in the intercellular spaces in the contact of the hyphae and the host cells (Fig. 5). Electron dense materials were also observed deposited around disorganized intracellular hyphae and in the vacuoles of colonized cells (Fig. 5c).

(TEM) Cork oak and holm oak roots pre-treated with α-CIN and infected with Phytophthora cinnamomi. a (cork oak) Intercellular and intracellular disorganized hyphae (P). Amorphous electron dense material (*) deposited around the intracellular hypha and along host cell vacuole. (HC = host cell). b, c and d (holm oak)- The pathogen (P) in the intercellular spaces (IS) of the cortex showing highly convoluted and disrupted plasmalemma (arrows), loss of membrane integrity (arrows) and of cytoplasm density, general cytoplasm disorganization and collapse. Electron dense materials (*) in the intercellular spaces in contact with hyphae (P)
Immunocytochemical localization of α-cinnamomin
α-cinnamomin secreted in the interaction Phytophthora cinnamomi-Quercus suber
Twenty four hpi of the cork oak roots with Phytophthora cinnamomi, α-CIN secreted by the pathogen was observed mainly at the periphery of the hyphae in the contact zone of P. cinnamomi with epidermal host cells and also in host cell walls, plasmalemma outer cytoplasm and around the intercellular hyphae in the vacuoles of invaded epidermal cells (Fig. 6).

(TEM) Immunocytochemical localization of cinnamomins secreted by P. cinnamomi 24 hpi of cork oak roots. a Contact zone of the pathogen (P) and an epidermal host cell (HC). The circle indicates the observed area in c and d. b Gold labelling in the contact region of the pathogen and host cell wall, on plasmalemma and outer cytoplasm of the host cell. c Gold labelling in a vacuole at the periphery of the pathogen. d Gold labelling around an intracellular hypha in the vacuole of an invaded epidermal cell
α-cinnamomin absorbed by cork oak roots
Alpha-cinnamomin absorbed by cork oak roots upon 24 h immersion was localized by the gold colloidal particles in the vacuoles of the cells of cortical and vascular parenchyma (Fig. 7).

(TEM) Immunocytochemical localization of α-CIN absorbed by cork oak roots after 24 h of immersion. a Localization (circle) of the observed zone of the vascular parenchyma presented in b. a and b Abundant osmiophylic materials (*) were seen bordering the vacuoles. b The immunogold labelling of α-CIN (arrows) was found in the cell vacuoles
Quantification of pathogen colonization
A quantitative real-time PCR (QPCR) assay following a method described by Eshraghi et al. (2011) was performed to study the effect of α-CIN and β-CIN on the colonization of Q. suber roots by P. cinnamomi. We assume that the proportion of pathogen and host biomass present at any given time during an infection is equivalent to the proportion of pathogen and plant DNA. Normalization of the pathogen DNA was established based on a purified plasmid DNA, containing the myelocytomatosis oncogene (Myc) of mouse as an internal control rather than host DNA to avoid any bias introduced by the variation of host DNA during Q. suber-P. cinnamomi interaction due to necrosis and degradation.
Plasmid and P. cinnamomi primers were found to be specific for their target DNA templates, Myc and PDN gene, respectively as described in M&M.
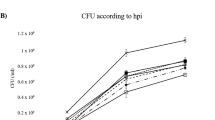
DNA levels of the pathogen in infected Q. suber roots relative to the internal plasmid were evaluated for 24 h through standard curves using QPCR. Root colonization by P. cinnamomi started 6 h after infection and gradually increased up to 24 h. Observations of distinct levels of P. cinnamomi infection in the cork oak roots during the infection period was shown to be reproducible. The major increase in P. cinnamomi/plasmid DNA ratio occurred between 18 and 24 hpi (Fig. 8).

Temporal colonization profile of Phytophthora cinnamomi in cork oak roots. Total qPCR quantification P. cinnamomi biomass over time, normalized based on plasmid DNA. Error bars represent the standard deviation of four biological replicates. The different letters indicate significant differences (P < 0.05)
The assay that was implemented for the quantitative measurement of P. cinnamomi DNA in Q. suber roots was applied to roots pre-treated with α-CIN and β-CIN in order to assess the elicitor effect of these cinnamomins in this pathosystem. As shown in Fig. 9, a significant reduction of P. cinnamomi infection in roots treated with 0.5 and 10 μM solutions of these cinnamomins was observed.

qPCR quantification of P. cinnamomi biomass in elicitin-treated and non-treated elicitin Quercus suber roots infected during 24 h at the shown concentrations. Different letters (a, b) indicate significant differences between treatments. Error bars represent the standard deviation of four biological replicates. The different letters indicate significant differences (P < 0.05)
Quantification of ROS
The production of H2O2 and O2 •− was measured at 6, 12, 18, 24, 36 and 48 h after infection with P. cinnamomi in 2-month-old cork oak roots with and without α-CIN treatment (Fig. 10a). The first significant level (P < 0.0001) of H2O2 peaked at 18 and 24 h in the infected elicitin non-treated plants. A 2.32-fold and 2.34-fold increase was observed at these time points, respectively, when compared with control plants (elicitin non-treated non-infected). In roots inoculated after treatment with α-CIN, H2O2 accumulation was detected earlier, at 12 h after infection, and was 1.85-fold higher than in elicitin-treated non-infected samples (P < 0.001); then, it increased slightly and peaked at 18 h (about 2-fold relative to the control; P < 0.0001) and showed a little decrease at 24 h (1.75-fold relative to the control; P < 0.01).

Kinetics of accumulation of reactive oxygen species (ROS) induced in cork oak roots submitted respectively to two treatments: inoculated with Phytophthora cinnamomi without (black graphs) or with a pre-treatment with α-CIN for 24 h (grey graphs). Non-inoculated plants in each treatment were used as a control. a H2O2 quantification. b O2 •− accumulation measured by XTT [sodium 3′ -[1-[phenylamino-carbonyl]-3,4-tetrazolium]-bis(4-methoxy-6-nitro) benzene-sulfonic acid hydrate] reduction. Statistically significant differences between infected and control plants for each treatment are indicated by different letters along each graph while asterisk shows significant differences between α-CIN pretreated and non-pretreated samples at each time point with P < 0.05. Error bars represent the standard deviation for three biological replicates
In non-treated elicitin inoculated roots, H2O2 followed a gradual decline from 24 to 36 h and then a statistically significant increase (P < 0.01) was observed at 48 hpi (1.89-fold relative to the control). This profile was not observed upon cinnamomin treatment whereby a continuous slower decrease was observed after 24 h. Moreover, at 18 and 24 h, a statistically significant difference (P < 0.05 and P < 0.001), corresponding to 1.16-fold and 1.32-fold higher H2O2 production, respectively, relative to the inoculated roots treated with cinnamomin, was observed.
The production of radical superoxide anion was evaluated at the same post-inoculation times. The pathogen induced the production of radical superoxide anion in the infected Q. suber roots (Fig. 10b). The O2 •− accumulation increased significantly during infection with the first significant (P < 0.001) augment observed at 24 h, corresponding to a 2-fold higher level of production relative to the control. It was followed by further accumulations at 36 and 48 h after infection, exhibiting 2.35-fold and about 3-fold increases (P < 0.0001), respectively as compared to the control. In roots pre-treated with α-CIN, the accumulation of radical superoxide anion exhibited a similar profile but with lower values: the first statistically significant (P < 0.05) O2 •− accumulation was detected at 24 h with a 1.56-fold increase relative to the control, and at 36 and 48 h after infection showed 2-fold and 2.57-fold increases (P < 0.0001), respectively.
Generally, elicitin pre-treated roots showed a lower level of O2 •− production during infection compared with the elicitin non-treated inoculated roots. This corresponds to a significant (P <0.05) decrease in O2 •− level of 1.49-fold and 1.30-fold at 24 and 48 h, respectively.
Activity of POD and ROS-scavenging enzymes
POD, SOD and CAT activities were determined in protein extracts from Q. suber roots during the time course of infection by P. cinnamomi with and without pre-treatment with α-CIN. Infection caused an increase in POD activity in Q. suber roots (Fig. 11a). In α-CIN non-treated roots, the first statistically significant (P < 0.05) increase started at 24 hpi (1.90-fold) and continuously increased during the period of observation: 2.27-fold and 3-fold at 36 and 48 hpi respectively, relative to the control samples (P < 0.001 and P < 0.0001, respectively). α-CIN pre-treated plants showed the first significant increase of POD activity at 18 hpi (about 2-fold relative to the control, P < 0.01) and continued to increase until 48 hpi [2.53-fold and 3.17-fold at 36 and 48 hpi, respectively (P < 0.0001)]. POD was induced at a higher level in roots pre-treated with elicitin when compared with those without pre-treatment. This was statistically significant at 24, 36 and 48 hpi (1.66-fold, 1.49-fold and 1.43-fold increases, with P < 0.05; P < 0.01; P < 0.05, respectively).

Determination of a) peroxidase (POD); b) superoxide dismutase (SOD) and c) catalase (CAT) activities in Quercus suber roots submitted respectively to two treatments: inoculated with Phytophthora cinnamomi without pre-treatment with α-CIN (black graphs) and inoculated with Phytophthora cinnamomi after pre-treatment with α-CIN for 24 h (grey graphs). Non-inoculated plants in each treatment were used as a control. Statistically significant differences (P < 0.05) between inoculated and control plants for each treatment are indicated with different letters along each graph. Asterisk means significant differences between α-CIN-pretreated and non-pretreated roots at the indicated time point with P < 0.05. Error bars represent the standard deviation for three biological replicates
In non-treated elicitin inoculated roots, the measurement of SOD activity showed a 1.80-fold and 1.92-fold significant (P < 0.0001) increase at 18 and 24 hpi in comparison to non-inoculated roots, then a decline until 48 h (Fig. 11b). In α-CIN pre-treated roots, SOD activity began to increase significantly at 12 hpi (1.53-fold higher relative to the control plants with P < 0.01), then followed a higher significant (P < 0.0001) increase at 18 and 24 hpi, (1.87-fold and 1.85-fold, respectively, relative to control plants) and a decrease from 24 to 48 hpi. Moreover, at 12 and at 18 hpi, 1.48 and 1.26-fold higher SOD activities, respectively were observed in roots pre-treated with α-CIN as compared with elicitin non-treated roots (P < 0.05).
The activity of catalase appears significantly increased at 36 and 48 hpi, in infected roots in the absence of elicitin pre-treatment (2.46 and 3.58-fold, respectively relative to the control group with P < 0.0001) whereas no significant change was observed before these time points (Fig. 11c). In roots treated with α-CIN, the catalase activity increased even more at 36 and 48 hpi. This differential increase of CAT activity showed to be significant (P < 0.05) at 18, 24 and 36 hpi with 1.59, 1.55 and 1.91-fold higher levels, respectively, in α-CIN pre-treated roots relative to elicitin non-treated roots.
Overall these results show that the α-CIN pre-treatment enhances the ROS-scavenging enzyme activity and also the total peroxidase activity in inoculated Q. suber roots.
Discussion
Despite numerous studies the role and mode of action of elicitins still remains largely unexplored. The present study reports for the first time the effects of cinnamomins on colonization of cork oak roots by P. cinnamomi, through biomass assessment, on the ROS species production and the enzymatic antioxidant activities upon infection, and on the localization of cinnamomins in root tissues. The present work also describes the cytological effects of α-CIN, an elicitin secreted by the pathogen itself, on cork oak and holm oak infection by P. cinnamomi.
Previous cytohistological observations have shown that P. cinnamomi colonizes Q. suber and Q. ilex penetrating to the phloem and xylem vessels and leading to occlusion, at least in 2- to 6-month-old plants (Pires et al. 2008; Horta et al. 2010; Medeira et al. 2012a; Ruiz-Gómez et al. 2014). In addition, recent cytological and physiological studies showed that cryptogein (elicitin secreted by Phytophthora cryptogea) and capsicein (secreted by Phytophthora capsici) induced a resistance response on cork oak against infection by P. cinnamomi (Medeira et al. 2012a) and that α-CIN has the same effect in Castanea sativa (Medeira et al. 2012b). In the present work we show that α-CIN induces a defence response to the infection by P. cinnamomi in Q. suber and in Q. ilex as well. The two oak species respond in a similar way as shown by the accumulation of electron-dense and of osmiophylic fibrillo-granular materials around disorganized intracellular hyphae which showed signs of cytoplasmic disorganization and, in all observed roots, were unable to reach and colonize the vascular cylinder, and by the restriction of the pathogen invasion to the cortex and to the intercellular spaces of the root, indicating an overall reduction of P. cinnamomi infection. The similarity of α–CIN effects in these two oak species with those observed for cryptogein and capsicein, and for oligandrin in tomato and grapevine challenged with Fusarium oxysporum and Botrytis cinerea, respectively (Benhamou et al. 2001; Mohamed et al. 2007) indicates that these elicitins share structural features responsible for their elicitor action and in addition, that an elicitin may preserve its elicitor capability when triggering a defence against the pathogen which produces it, as shown in this work for the binomial α-CIN - P. cinnamomi.
The ability of elicitins to induce a defence reaction in Q. suber was further evidenced using an approach to measure the pathogen biomass in roots, based on the accurate quantification of P. cinnamomi DNA by QPCR. This assay, developed by Eshraghi et al. (2011), was applied first to establish a temporal colonization profile of cork oak roots which showed a highly significant colonization rate between 18 and 24 hpi. For reasons of availability, we used as internal control a plasmid containing the myelocytomatosis oncogene (Myc) of mouse, instead of the mouse ScFvB1sequence used by the authors. It also has no significant homology in known plant and fungal genomes, and did not interfere with the amplification of the pathogen DNA. To assess the effect of elicitins on the colonization of roots, said roots were pre-treated with two concentrations of the two main P. cinnamomi elicitins, α-CIN and β-CIN and the quantification of the pathogen was carried out at 24 hpi. At a concentration, as low as 0.5 μM of each elicitin, a decrease of 3- to 1.8-fold in the amount of the pathogen was observed. There were no significant differences concerning either the nature of the elicitin or the concentration from 0.5 to 10 μM. These findings suggest that the physico-chemical differences between these two elicitins, in particular their pI (4.4 for the acidic α-CIN, 8.9 for the basic β-CIN), do not appear to influence their capacity to reduce the pathogen biomass. This reduction in the pathogen infection after cinnamomin treatment can be interpreted as an induced defence reaction and corroborates the cytohistological findings.
Using the immunogold cytolocalization technique we showed that cinnamomin secreted by P. cinnamomi in 24 h interaction with cork and holm oak, was mainly present in the walls and outer cytoplasm of the hyphae but was also visible in the cell walls, outer cytoplasm and vacuoles of the epidermal cells of the host. This observation is compatible with known actions of elicitins, namely the metabolic cascade of events involving plasma membrane functions following elicitin recognition by cell plasma membrane (Pugin and Guern 1996) and with the transport of captured sterols to the plasma membrane, as their size enables them to pass freely across the cell wall [as canonical elicitins are sterol carrier proteins (Mikes et al. 1997)]. Their presence in vacuoles could be the way to their isolation and elimination after being recognized as a threat by the host cell. In the present work we did not intend to quantify elicitins in root tissue and results are not fully comparable with those reported by Brummer et al. (2002) but, except for the localization in vacuoles, these observations agree with those concerning the quercinin in the interaction of Phytophthora quercina with Quercus robur roots. It is not expected that polyclonal antibodies used to prepare the gold immune probe are specific to α-CIN; thus, the observed stained particles correspond very probably to the different classes of cinnamomi secreted by P. cinnamomin, although α-CIN is predictably the most abundant (Horta et al. 2008).
The same method was used to localize the elicitin upon immersion of cork oak roots in an aqueous solution of α-CIN. The observed migration of the elicitin towards the vascular cylinder suggests its transport to other plant organs to initiate a general defence mechanism.
The production ROS in the so-called oxidative burst is one of the manifestations of defence responses against fungal and oomycete pathogens. The characteristic times of ROS production for pathogenic infections may vary with distinct biological species. Information on the involvement of ROS accumulation during Phytophthora colonization is lacking for Quercus. In the present study we focused on ROS production during the early stages of the interaction (0–48 h) between P. cinnamomi and Q. suber. H2O2 peaked at 18 and 24 h in the infected roots in close agreement with the observed temporal colonization profile of P. cinnamomi. The pathogen induced the production of radical superoxide anion as well, with the first significant increase observed at 24 h, but it was followed by further accumulations at 36 and 48 h after infection. The production of ROS in these interactions can debilitate the host tissue at early stages of infection and, consequently enhance growth and development of the pathogen. In fact, P. cinnamomi is a hemi-biotroph pathogen that can also colonize dead tissue and could therefore benefit from such a host defence response and higher ROS accumulation as demonstrated in the interaction of avocado roots and P. cinnamomi ( García-Pineda et al. 2010). In addition, other studies have shown that higher ROS accumulation results in greater susceptibility to necrotrophic fungi infection, whereas a reduced ROS level confers higher disease resistance (van Kan 2006). Pre-treatment with α-CIN resulted in a significant reduction of ROS accumulation in infected roots, during all the time course of infection for O2 •− and namely at times 18 and 24 hpi for H2O2, when its production is peaked. Therefore, a lower level of ROS production after α-CIN treatment suggests the elicitin exerts a protective effect through a reduction of host cell damage.
Although relative high levels of ROS can be toxic, lower levels may serve as signaling molecules involved in regulation of defence responses (Lamb and Dixon 1997). They can behave, for instance as a second messenger participating in different cellular pathways including tolerance to environmental stresses in the host-pathogen interaction. As ROS are important mediators of various functions in plants, their regulation is essential for the cells to avoid any oxidative injury and not to remove them entirely. SOD and CAT are examples of major ROS scavenging enzymes produced to avoid cellular disintegration caused by ROS. Suppressing ROS-scavenging systems leads to the inability of cells to dismutate O2 • ─ and decompose H2O2 and to over-accumulation of ROS (reviewed by Mittler 2002).
In this study we monitored enzymes that regulate oxidative species measuring SOD, CAT and POD activities. The SOD activity in infected cork oak roots increased markedly when compared with the control suggesting an important role of this enzyme in coping with ROS produced by cork oak roots during plant infection. In the Quercus suber–P. cinnamomi pathosystem, the increase in SOD activity was coincident with the more significant H2O2 accumulation that occurred at 18 and 24 hpi suggesting that increases in SOD activity could contribute to the observed high levels of H2O2. Furthermore, in the present study, the SOD activity increased in roots pre-treated with α-CIN before inoculation when compared with the corresponding control, following the same time course pattern. The results are in accordance with those obtained by Sahoo et al. (2007) who reported that the activity of SOD increased in taro (Colocasia esculenta) under induced blight condition with P. colocasiae and that this increase was higher in resistance genotypes when compared with susceptible ones. The role of SOD during plant infection by pathogenic organisms appears to be the dismutation of O2 •−into H2O2 produced by plasmalemma -bound nicotinamide adenine dinucleotide phosphate oxidases, cell-wall-bound POXs and amine oxidases (Mittler 2002). To decompose H2O2 in non-ROS, CAT is required and its action must be coordinated with SOD to attenuate ROS accumulation and mitigate their destructive action that could result from the imbalanced activities of the enzymes. Coincident with a high significant increase in SOD activity observed in cork oak roots 18 h after P. cinnamomi inoculation, the basal CAT activity could explain the H2O2 overproduction. The subsequent increase in CAT activity 36–48 hpi could reflect the ability of H2O2 scavenging in this pathosystem.
CAT is the most effective scavenging enzyme in preventing oxidative damage (Mittler 2002). In the present work the CAT activity was stimulated significantly upon pre-treatment with α-CIN when compared with the control group assuring a more effective contribution to the reduction of H2O2 content. The activation of CAT is an important contribution to the protection of host plant roots against overproduction of H2O2.
All together these observations suggest there is a balanced activity of SOD and CAT to cope with ROS production in the Quercus suber-P. cinnamomi pathosystem and that α-CIN pre-treatment of host roots triggers a defence response through the increasing of the activities of the enzymes.
Peroxidases participate in the phenylpropanoid metabolism whereby lignin and suberin are produced. POD uses H2O2 as the ultimate electron acceptor to catalyse the final step in the synthesis of lignin from the oxidation of cinnamyl alcohols. Lignification of plant cell walls is a manifestation of a defence reaction acting as a physical barrier that blocks the entry of pathogens (Vance et al. 1980). In this study, POD activity was found to be higher in infected plants in comparison with the healthy ones. Similar results were observed on POD activity in avocado roots infected with P. cinnamomi (García-Pineda et al. 2010). In cork oak roots the increase in enzyme activity start to be significant from 24 hpi when compared with controls, coincident with one peak of H2O2 production. In α-CIN pre-treated roots the activity of POD underwent a further remarkable increase when compared with the corresponding control. Similar results were observed on POD activity induction in tomato plants treated with oligandrin after inoculation with B. cinerea in comparison with infected plants without treatment (Lou et al. 2011). Peroxidase also generates free radicals and H2O2 which can participate in a response defence against pathogen activity as discussed above. It was out of the scope of this work to ascertain lignin and phenol contents. However, cinnamyl alcohol dehydrogenase (CAD) genes have been shown to be up-regulated in Q. suber root seedlings infected with P. cinnamomi (Coelho et al. 2006; Ebadzad and Cravador 2014) suggesting that the observed POD activity increase is related with the participation of this enzyme in the oxidation of the CADs.
The understanding of specific Quercus-P. cinnamomi interactions involves the implementation of multidisciplinary approaches directed to the unravelling of the mechanisms that control pathogen invasion and host resistance responses. In this study, measuring pathogen biomass, ROS production and antioxidant enzyme activities in Q. suber roots treated with elicitins, and performing histopathological studies in cork and holm oak allowed a complementary insight into the effect of elicitins on P. cinnamomi infection in these oaks.
In conclusion, this feature of elicitins being able to trigger defence responses is more general than it was first believed (Grant et al. 1996; Ponchet et al. 1999). This property may be assumed, without detriment to other demonstrated roles such as being essential players in the colonization process and being sterol transporters. Their action is not genus- or cultivar-specific and not only elicitins from Phytophthora but also from Pythium, like oligandrin play this role. Plants in the family Fagaceae as shown here and in other reports (Medeira et al. 2012a and Medeira et al. 2012b) using various approaches, display defence responses induced by elicitins.
The defence triggering activity of cinnamomins shown in the present work appears to conflict with their role as a virulence factor, as demonstrated in the interaction Q. suber- P. cinnamomi (Horta et al. 2010). This dual conflicting activity has been reported before in various plant-microbial pathosystems (Van’t Slot and Knogge 2002). The term effectors was coined to designate molecules capable of manipulating host cell structure and function, thereby facilitating infection or triggering defense responses (Kamoun 2006). We hypothesize that when host cells are exposed to cinnamomins prior to challenge with the oomycete, these molecules behave mainly as apoplastic effectors inducing primary defense mechanisms mediated by receptors involved in their recognition. In absence of prior exposition to elicitin, cinnamomins translocated by the pathogen into the host cell act as cytoplasmic effectors, disrupting the activation and execution of plant defenses by an unknown mechanism.
Whether the defence effect triggered by cinnamomins is persistent enough to be usefully exploited remains to be shown and assuredly will implies the study of molecular receptors involved in the recognition of these effectors and in enhanced resistance/tolerance to P. cinnamomi.
References
Able, A. J., Guest, D. I., & Sutherland, M. W. (1998). Use of a new tetrazolium-based assay to study the production of superoxide radicals by tobacco cell cultures challenged with avirulent zoospores of Phytophthora parasitica var nicotianae. Plant Physiology, 117, 491–499.
Aebi, H. (1984). Catalase in vitro. Methods in Enzymology, 105, 121–126.
Apel, K., & Hirt, H. (2004). Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annual Reveiw Plant Biology, 55, 373–399.
Archer, M., Rodrigues, M. L., Aurelio, M., Biemans, R., Cravador, A., & Carrondo, M. A. (2000). Acta Crystallographica, D56, 363–365.
Benhamou, N., Bélanger, R. R., Rey, P., & Tirilly, Y. (2001). Oligandrin, the elicitin like protein produced by the mycoparasite Pythium oligandrum, induces systemic resistance to Fusarium crown and root rot in tomato plants. Plant Physiology and Biochemistry, 39, 681–698.
Beyer, W. F., & Fridovich, I. (1987). Assaying for superoxide dismutase activity: some large consequences of minor changes in conditions. Analitical Biochemistry, 161, 559–566.
Blein, J. P., Coutos-Thévenot, P., Marion, D., & Ponchet, M. (2002). From elicitins to lipid-transfer proteins: a new insight in cell signalling involved in plant defense mechanisms. Trends Plant Science, 7, 293–296.
Bonnet, P., Bourdon, E., Ponchet, M., Blein, J.-P., & Ricci, P. (1996). Acquired resistance triggered by elicitins in tobacco and other plants. European Journal of Plant Pathology, 102, 181–192.
Brasier, C. M., Robredo, F., & Ferraz, J. F. P. (1993). Evidence for Phytophthora cinnamomi involvement in Iberian oak decline. Plant Pathology, 42, 140–145.
Brummer, M., Arend, M., Fromm, J., Schlenzig, A., & Oβwald, W. F. (2002). Ultrastructural changes and immunocytochemical localization of the elicitin quercinin in Quercus robur L. roots infected with Phytophthora quercina. Physiological and Molecular Plant Pathology, 61, 109–120.
Caetano, P. (2007). Envolvimento de Phytophthora cinnamomi no declínio de Quercus suber e Q. rotundifolia: estudo da influência de factores bióticos e abióticos na progressão da doença. Possibilidades de controlo químico do declínio. PhD Dissertation, Universidade do Algarve, Portugal. http://sapientia.ualg.pt/handle/10400.1/400
Cakmak, I., & Marschner, H. (1992). Magnesium deficiency and high light intensity enhance activities of superoxide dismutase, ascorbate peroxidase, and glutathione reductase in bean leaves. Plant Physiology, 98, 1222–1227.
Coelho, A. C., Horta, M., Neves, D., & Cravador, A. (2006). Involvement of a cinnamyl alcohol dehydrogenase of Quercus suber in the defence response to infection by Phytophthora cinnamomi. Physiological and Molecular Plant Pathology, 69, 62–72.
Crandall, B. S. (1950). The distribution and significance of the chestnut root rot Phytophthoras, P. cinnamomi and P. cambivora. Plant Disease Reporter, 34, 194–6.
Ebadzad, G., & Cravador, A. (2014). Quantitative RT-PCR analysis of differentially expressed genes in Quercus suber in response to Phytophthora cinnamomi infection. Springer Plus, 3, 613. doi:10.1186/2193-1801-3-613.
Eshraghi, L., Aryamanesh, N., Anderson, J. P., Shearer, B., McComb, J. A., Hardy, G. E. S. J., & O’Brien, P. A. (2011). A quantitative PCR assay for accurate in planta quantification of the necrotrophic pathogen Phytophthora cinnamomi. European Journal of Plant Pathology, 131, 419–430.
García-Pineda, E., Benezer-Benezer, M., Gutiérrez-Segundo, A., Rangel-Sánchez, G., Arreola-Cortés, A., & Castro-Mercado, E. (2010). Regulation of defence responses in avocado roots infected with Phytophthora cinnamomi. Plant and Soil, 331, 45–56.
Grant, R. B., Ebert, D., & Gayler, K. R. (1996). Elicitins: proteins in search of a role? Autralasian Plant Pathology, 25, 148–157.
Horta, M., Sousa, N., Coelho, A. C., Neves, D., & Cravador, A. (2008). In vitro and in vivo quantification of elicitin expression in Phytophthora cinnamomi. Physiological and Molecular Plant Pathology, 73, 48–57.
Horta, M., Caetano, P., Coelho, A. C., Medeira, C., Maia, I., Neves, D., & Cravador, A. (2010). Involvement of the β-cinnamomin elicitin in infection and colonisation of cork oak roots by Phytophthora cinnamomi. European Journal of Plant Pathology, 127, 427–436. doi:10.1007/s10658-010-9609-x.
Kamoun, S. (2006). A catalogue of the effector secretome of plant pathogenic oomycetes. Annual Review of Phytopathology, 44, 41–60.
Lamb, C., & Dixon, R. A. (1997). The oxidative burst in plant disease resistance. Annual Review of Plant Pathology and Plant Molecular Biology, 48, 251–275.
Lou, B., Wan, A., Lin, C. H., Xu, T., & Zhen, X. (2011). Enhancement of defence responses by oligandrin against Botrytis cinerea in tomatoes. African Journal of Biotechnology, 10(55), 11442–11449.
Manter, D. K., Kolodny, E. H., Hansen, E. M., & Parke, J. L. (2010). Virulence, sporulation and elicitin production in three clonal lineages of Phytophthora ramorum. Physiological and Molecular Plant Pathology, 74, 317–322.
Medeira, C., Quartin, V., Maia, I., Diniz, I., Matos, M. C., Semedo, J., Scotti-Campos, P., Ramalho, J., Pais, I., Ramos, P., Melo, E., Leitão, A., & Cravador, A. (2012a). Cryptogein and capsicein promote defence responses in Quercus suber against Phytophthora cinnamomi infection. European Journal of Plant Pathology, 134, 145–159. doi:10.1007/s10658-012-9972-x.
Medeira, C., Maia, I., Ribeiro, C., Candeias, I., Melo, E., Sousa, N., & Cravador, A. (2012b). Alpha cinnamomin elicit a defence response against Phytophthora cinnamomi in Castanea sativa. ISHS Acta Horticulturae, 940, 315–320.
Mikes, V., Milat, M.-L., Ponchet, M., Ricci, P., & Blein, J.-P. (1997). The fungal elicitor cryptogein is a sterol carrier protein. FEBS Letters, 416, 190–192.
Mittler, R. (2002). Oxidative stress, antioxidants and stress tolerance. Trends in Plant Science, 7, 405–410.
Mohamed, N., Lheirminier, J., Farmer, M.-J., Fromentin, J., Béno, N., Houot, V., Milat, M.-L., & Blein, J. P. (2007). Defense responses in grape vine leaves against Botrytris cinerea induced by application of a Pythium oligandrum strain or its elicitin, oligandrin, to roots. Phytopatology, 97, 611–620.
Oßwald, W., Fleischmann, F., Rigling, D., Coelho, A. C., Cravador, A., Diez, J., Dalio, R. J., Horta Jung, M., Pfanz, H., Robin, C., Sipos, G., Solla, A., Cech, T., Chambery, A., Diamandis, S., Hansen, E., Jung, T., Orlikowski, L. B., Parke, J., Prospero, S., & Werres, S. (2014). Strategies of attack and defence in woody plant-Phytophthora interactions. Forest Pathology, 44(3), 169–190. doi:10.1111/efp.12096.
Pires, N., Maia, I., Moreira, A., & Medeira, C. (2008). Early stages of infection of cork and holm oak trees by Phytophthora cinnamomi. In J. Vázquez & H. Pereira (Eds.), Suberwood: New challenges for the integration of cork oak forests and products (pp. 275–282). Spain: Universidad de Huelva.
Ponchet, M., Panabières, F., Milat, M. L., Mikes, V., Montillet, J. L., Suty, L., Triantaphylides, C., Tirilly, Y., & Blein, J. P. (1999). Are the elicitins cryptograms in plant-oomycete communications? Cellular and Molecular Life Sciences, 56, 1020–1047.
Pugin, A., & Guern, J. (1996). Mode of action of elicitors: involvement of plasma membrane functions. Comptes Rendus de l’Académie des Sciences, Série, 3(319), 1055–1061.
Ricci, P., Bonnet, P., Huet, J. C., Sallantin, M., Beauvais-Cante, F., Bruneteau, M., Billard, V., Michel, G., & Pernollet, J. C. (1989). Structure and activity of proteins from pathogenic fungi Phytophthora eliciting necrosis and acquired resistance in tobacco. European Journal of Biochemistry, 183, 555–563.
Robin, C., Desprez-Loustau, M.-L., Capron, G., & Delatour, C. (1998). First record of Phytophthora cinnamomi on cork and holm oaks in France and evidence of pathogenicity. Annual of Forest Science, 55, 869–883.
Rodrigues, M. L., Archer, M., Martel, P., Miranda, S., Thomaz, M., Enguita, F. J., Baptista, R. P., Melo, E. P., Sousa, N., Cravador, A., & Carrondo, M. A. (2006). Crystal structures of the free and sterol-bound forms of beta-cinnamomin. BBA-Proteins. Proteomics., 1764, 110–121.
Ruiz-Gómez, F. J., Navarro-Cerrillo, R. M., Sánchez-Cuesta, R., & Pérez-de-Luque, A. (2014). Histopathology of infection and colonization of Quercus ilex fine roots by Phytophthora cinnamomi. Plant Pathology. doi:10.1111/ppa.12310.
Sahoo, M. R., Das Gupta, M., Kole, P. C., Bhat, J. S., & Mukherjee, A. (2007). Antioxidative enzymes and isozymes analysis of taro genotypes and their implications in Phytophthora blight disease resistance. Mycopathologia, 163, 241–248.
Sánchez, M. E., Caetano, P., Ferraz, J., & Trapero, A. (2002). Phytophthora disease of Quercus ilex in south-western Spain. Forest Pathology, 32, 5–18.
Shearer, B. L., & Tippett, J. T. (1989). Jarrah Dieback: the dynamics and management of P. cinnamomi in the Jarrah (Eucalyptus marginata) Forest of South-western Australia. Department of conservation and land management. Western Australia: Research Bulletin, 3, 1–76.
van Kan, J. A. (2006). Licensed to kill: the lifestyle of a necrotrophic plant pathogen. Trends Plant Science, 118(1), 247–253.
Van’t Slot, K. A. E., & Knogge, W. (2002). A dual role for microbial pathogen-derived effector proteins in plant disease and resistance. Critical Reviews in Plant Sciences, 21, 229–271.
Vance, C. P., Kirk, T. K., & Sherwood, R. T. (1980). Lignification as a mechanism of disease resistance. Annual Revivew Phytopathology, 18, 259–288.
Acknowledgments
This work was financed by the Portuguese Ministério da Ciência e do Ensino Superior (MCES) (PTCD/AGR-AAM/68628/2006). Ghazal Ebadzad thanks Fundação para a Ciência e a Tecnologia (FCT) for her grant (SFRH/BD/76979/2011) and Erasmus Mundus (EM8) program. The manuscript does not infringe any other person’s copyright or property rights.
Conflict of interest
The authors declare no conflicts of interest.
Compliance with ethical standards
The present research did not involve either animals or human participants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ebadzad, G., Medeira, C., Maia, I. et al. Induction of defence responses by cinnamomins against Phytophthora cinnamomi in Quercus suber and Quercus ilex subs. rotundifolia . Eur J Plant Pathol 143, 705–723 (2015). https://doi.org/10.1007/s10658-015-0721-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-015-0721-9