
Abstract
Potato early blight and brown spot are important fungal diseases responsible for premature defoliation and yield loss of potato. Pathogens considered to be involved in leaf necrosis are Alternaria solani and A. alternata, respectively. Both diseases are commonly characterized by the visualization of leaf lesions. Current detection and identification methods for Alternaria species rely primarily on cultural and morphological characteristics, the assessment of which is time-consuming and not always suitable. Sensitive, reliable methods for estimating infection severity are therefore desirable. In this study, an Alternaria-specific real-time PCR assay was developed using primers based on internal transcribed spacers (ITS) 1 and 2. The assays facilitated species detection and clearly discriminated between A. solani and A. alternata. The use of real-time PCR allowed quantitative estimation of fungal biomass in plant tissues. Detection sensitivities were in the range of >100 fg. Real-time PCR applications used to accurately assess the extent of colonization by Alternaria spp. during disease development are reported here for the first time. Additionally, Alternaria genomic DNA levels were verified not only in potato leaves showing different levels of disease progress, but also in symptomless leaves. This assay provides a useful tool to quantify pathogen levels during initial latent stages of infection and will thus help in the early detection and quantification of Alternaria spp..
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Potato early blight and brown spot are known as important diseases of potato (Solanum tuberosum L.). The two closely related diseases are caused by fungi from the same genus. Early blight (EB) is caused by Alternaria solani (Ellis & G. Martin) L.R. Jones while brown spot lesions are caused by Alternaria alternata (Fr.) Keissl.. Both Alternaria species can reside on plant debris as well as directly in the soil. They represent a risk to crop productivity and can cause considerable yield loss. EB has been recognized as more important because it is highly destructive (Rotem 1994), and can be found in many potato growing regions worldwide. The economic concern associated with EB is well documented (Rotem 1994; van der Waals et al. 2003; Leiminger and Hausladen 2012). In recent years, EB has become increasingly important for European countries (Hausladen and Leiminger 2007), and significant yield losses up to 25 % have been reported due to premature death of the plants (Hausladen and Leiminger 2007). According to the literature, EB is seen as the predominant causal agent leading to symptoms including characteristic “target-like” lesions of concentric rings that appear dark, sunken and become papery (Rotem 1994; van der Waals et al. 2003). Brown spot, caused by A. alternata, tends to cause smaller leaf lesions that are usually darker in colour than EB lesions. However, in some cases brown spot can be highly similar in appearance and behaviour to EB causing potential confusion, as it may form similar lesions (Boiteux and Reifschneider 1989). Similarly, several studies have shown that A. solani and A. alternata can be isolated simultaneously from typical EB lesions (Bäßler et al. 2004; Latorse et al. 2010), preventing a clear differentiation in the field. Rotem (1994) stated A. alternata as a secondary parasite, which may appear in up to 25 % of lesions initiated by A. solani.
Up to now, techniques for Alternaria identification have relied mainly on agar plate methods, where Alternaria species are differentiated microscopically by spore morphology. Latorse et al. (2010) reported that A. alternata differentiates spores much faster than A. solani when isolated on artificial medium, introducing a bias in this morphological diagnostic. Similarly, a quantitative estimation of fungal biomass in plant tissue cannot be achieved, although the quantification of pathogen biomass in planta is a crucial step when monitoring disease epidemiology. Accordingly, agar plate methods do not guarantee a distinct differentiation of pathogens. In the field, disease scoring is widely used to quantify disease progress (Granovsky and Peterson 1954). However, this method also does not guarantee a distinct differentiation of pathogens, as it is only based on the visual assessment of symptoms. Additionally, monitoring of latent infection is not possible. The most commonly used molecular technique for the identification of Alternaria species is conventional PCR with species-specific primers based on the ribosomal DNA internal transcribed spacer (ITS) (Guillemette et al. 2004). Various methods have been developed for the detection and identification of Alternaria spp. in cereals and vegetables (Konstantinova et al. 2002; Zur et al. 2002). However, when the quantity of target DNA is very small, the sensitivity of conventional PCR is inadequate. Further molecular techniques are needed to guarantee rapid and reliable pathogen quantification. Here, quantitative polymerase chain reaction (real-time PCR) can be helpful to quantify and differentiate pathogens because of its increased sensitivity (Schena et al. 2004; Pavón et al. 2010). Since its development in the 1990s, real-time PCR has emerged as a reliable and sensitive method for detecting and quantifying phytopathogenic and antagonistic fungi (Schena et al. 2004), and represents a highly sensitive and specific technique for the detection and quantification of nucleic acids (Tylor et al. 2001). Protocols for the qualitative recognition of Alternaria species in sunflowers and foodstuff have recently been established (Pavón et al. 2012; Udayashankar et al. 2012). Quantitative detection of A. alternata in tomato has been developed by Schuhegger et al. (2006).
In this study we describe real-time PCR applications to accurately assess the extent of Alternaria colonization of potato leaves during early disease development, and to differentiate the two species in question. To our knowledge, quantitative analyses in field studies assessing disease progression have not yet been performed. The use of this assay allows improved diagnostics of both Alternaria species as well as tracking the specific progression of Alternaria species within the host.
Materials and methods
Field studies
Leaf samples were taken from naturally infected potato fields. Fields were located in Freising-Weihenstephan and Schwaig (Bavaria) in 2003 and 2004, and in Straubing (Bavaria) from 2005 to 2007. Sampling schemes were designed as a randomized complete block and were replicated four times. Field plots consisted of four rows 8 m long and 0.75 m between rows (24 m2). Plant density was 40,000 plants ha−1. Tests were carried out using the potato cultivars Karlena (2003) and Kuras (2004 to 2007), both of which are susceptible to EB. Experimental plots were situated within a commercial potato field which remained non-treated against EB, thus providing natural inocula for EB disease development. All plots were fertilized and cultivated according to general agricultural practice.
Visual evaluation of EB infection
Disease progression was observed weekly from emergence until death of the potato plants. Within each of the replications, 10 plants per plot were monitored for disease progression of EB or other diseases (e.g. late blight). In 2003 and 2004, the percentage of EB-infected leaf area was assessed visually by observing the middle section of potato plants. In order to follow up disease development in more detail, potato plants were divided into three leaf layers (bottom, middle and upper leaf layer) from 2005 onwards. The percentage of EB-infected leaf area was assessed visually for each leaf layer of the potato plants according to Granovsky and Peterson (1954). Leaf symptoms were not assigned to either A. solani or A. alternata but were, for the sake of simplicity, rated as total EB infections. The disease severity per plant was calculated as a mean value. The disease evaluation was omitted when leaves were either completely dead or when the lower leaves had senesced and dropped from the plant.
Fungicide treatments
To prevent late blight disease, potatoes were treated with the fungicide Ranman (cyazofamid at 400 g l−1) at a dose of 0.2 l ha−1 every 8 to 10 days. All plots were completely protected by cover sprays. In 2005 and 2006, plots treated weekly with mancozeb (1350 g a.i. ha−1) or azoxystrobin (125 g a.i. ha−1) were included. The parallel investigation of non-treated and treated plots allowed the investigation of how far pathogen development could be retarded by fungicide treatments.
Collection of plant material
From the onset of first symptoms, which were initially found at the bottom leaf layer, leaf samples were taken approx. every 14 days from EB non-treated control plots. In 2003 and 2004, leaves were randomly collected from the middle section of potato plants. Each plot was divided into four subplots. Ten leaves were taken for each subplot, which were mixed for further analysis. In 2005 to 2007 potato plants were separated into individual leaf layers (bottom, middle and upper leaf layer) to follow up EB disease development. The sampling process itself was identical to the previous years, and 10 leaves were collected for each leaf layer. Tissue was harvested and immediately frozen in liquid nitrogen at the site. For leaf levels which did not contain any leaflets due to premature defoliation caused by EB, sampling was discontinued. In 2005 and 2006, leaf samples were simultaneously taken from EB non-treated plots and plots treated weekly with either mancozeb (1350 g a.i. ha−1) or azoxystrobin (125 g a.i. ha−1). The parallel investigation of non-treated and treated leaf samples was carried out in order to determine if molecular analysis can be used to monitor disease control. Samples were taken from the middle and upper leaf sections at one sampling date at the end of the season (sampling date 2005: Aug 12th, 2006: Sept. 1st). Each plot was repeated four times and each repetition was divided into four subplots. From each subplot 10 leaves were sampled and homogenized to one sample.
Genomic DNA extraction
Samples were carefully ground in liquid nitrogen and crushed with a pestle to a fine powder. Samples were homogenized and aliquots of 100 mg were used in lieu of fresh weight according to Bahnweg et al. (1998). The final DNA was dissolved in 100 μl Tris EDTA (TE buffer, pH 8) and adjusted to 25 ng μl−1. Similarly, DNA was also extracted from reference strains of A. solani and A. alternata.
PCR primer development
Real-time PCR analysis was carried out to elucidate which of the two Alternaria species was dominating on a potato leaf bearing symptoms. ITS regions of three isolates of Alternaria solani and three isolates of A. alternata (obtained from CBS, Utrecht, The Netherlands and from DSMZ, Braunschweig, Germany) were sequenced. The sequences spanning the ITS1, ITS2 regions and the 5.8 S genes were aligned using DiAlign (Genomatix, Munich, Germany). Sequences were identified which were 18–22 bases in length and present in one species, but not the other. Sequence pairs, preferably around 100–350 bases apart from each other, were selected for primer synthesis. The forward primer was identical to the base sequence obtained by sequencing, whilst the reverse primer was the reverse complement sequence. Rt-primers used for the quantification of A. alternata have been previously developed by Schuhegger et al. (2006). The PCR product size was 96 bp). ITS base sequences of A. solani had been determined from commercially available strains (Centraalbureau voor Schimmelcultures, Utrecht, The Netherlands). One microlitre of the extracted DNA solution (adjusted to 25 ng μl−1) was used for real-time PCR which was performed in 25 μl reactions. The designed primers for A. solani were AsFor, 5’ TCCGTAGCTGAACCTGCGG 3’ and AsRev, 5’ TGGGTTGGTCCTTGTGGTG 3’. Quantitative PCR of A. solani produced a 152 bp DNA fragment. No amplicon was obtained from healthy potato leaves. Validity of positive amplification signals was routinely checked by determining melting curves of the amplification products.
Real-time PCR specificity and sensitivity
DNA extracted from different isolates of A. solani and A. alternata was used as a template for PCR amplification under the conditions described above in order to determine the species specificity of the primers. The specificity of the qPCR system was verified by analysis of the DNA extracted from a culture collection (86 isolates) of 48 fungi pathogenic to species of the Solanaceae (Table 1).
Standard curve preparation
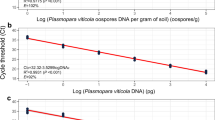
The sensitivity of both primers was assessed using serial dilutions of purified genomic DNA. A standard curve was generated for each run with a dilution series of purified and calibrated A. solani and A. alternata genomic DNAs (200 ng μl−1 stock). Standard curves were obtained using repetitive 10-fold dilutions, with 1:10 being equal to 20 ng fungal DNA, 1:100 equal to 2 ng fungal DNA etc., down to 2 pg fungal DNA. The standard curve was developed by plotting the logarithm of known concentrations of A. solani and A. alternata DNA against the quantification cycle (Cq) values (Fig. 1). Cq can be defined as the fractional cycle number at which the fluorescence of the PCR product is significantly different from the background. Cq value is inversely related to log of initial concentration; therefore, the lower the Cq value, the higher the initial DNA concentration.

Confidence bands for calibration curve options. The x axis represents the Cq value for each calibration point (log copies mL−1) and the y axis the calibration curve quantity
PCR amplification and quantification
The real-time PCR reaction mixture contained the following components: 10 μl SYBR Green® Low ROX Mix Green Master Mix (AB Gene, Hamburg, Germany), including PCR buffer, Taq DNA polymerase and dNTPs, 0.5 μl of each primer and PCR-grade water to make up to a final volume of 25 μl. PCR amplification was performed in a PCR Real-Time System (ABI PRISM 7700, Applied Biosystems, Darmstadt, Germany). Thermocycler conditions were 94 °C for 15 min (activation of enzyme), 40 cycles of 94 °C for 15 s, 60 °C for 1 min (denaturation, annealing and amplification, respectively). The newly generated double-stranded DNA amplicon was quantified measuring the SYBR Green fluorescence intensity after each 60 °C step of the cycler program. Analysis of experiments (i.e. determination of Cq (quantification cycle) values to quantify initial amounts of target DNA) was conducted using the software 7500 Fast System v. 1.3.1. (Applied Biosystems). Validity of positive amplification signals was routinely checked by determining melting curves of the amplification products. In order to allow the processing of large sample numbers PCR assay conditions for the quantification of both A. solani and A. alternata were adapted to the same conditions.
Data analysis
Statistical analysis was carried out using the software package SPSS (SPSS Inc.). DNA values were submitted to analysis of variance. Mean values were compared using the Tukey test. The level of probability used in all statistical tests was P < 0.05.
Results
Visual quantification of early blight disease development and monitoring Alternaria development in plant tissue
The increase in disease severity caused by Alternaria spp. was monitored by visual disease scorings. Disease ratings revealed that first disease symptoms occurred at a very young age in potato plants. Throughout the study period, first symptoms were observed 3 to 6 weeks after crop emergence (Fig. 2). Here, EB was the primary foliar disease among all observed diseases. First disease symptoms appeared primarily on old leaves and were the size of pin heads, making them inconspicuous and therefore difficult to detect visually. Over all years, disease epidemics progressed slowly until the end of July, and more rapidly thereafter (Fig. 2). As the season progressed, EB symptoms rapidly increased and spread onto higher leaf levels. Secondary spread of EB was observed in all years starting at the end of July or the beginning of August. Heavy EB epidemics occurred throughout all 5 years of investigation. However, favourable weather conditions, together with the use of the early maturing cultivar Karlena, led to an earlier increase in disease epidemic in 2003 compared to 2004–2007. Here, potato plants were totally destroyed by EB by the end of August. In 2004–2006, EB resulted in leaf death in mid-September. In 2007, increase in disease epidemic was observed 14 days earlier than in 2006 and potato plants had been destroyed by the beginning of September. In parallel to this, the development and accumulation of mycelial growth within the plant tissue was estimated by real-time PCR analysis.

Disease progression of early blight (EB) between 2003 and 2007 at locations at Freising (2003), Schwaig (2004) and Straubing (2005–2007), Germany. In 2003 the potato cultivar Karlena was planted, whereas in 2004 to 2007 cultivation was carried out with cultivar Kuras. To prevent late blight disease, potatoes were treated with the fungicide Ranman (cyazofamid at 400 g l−1) at a dose of 0.2 l ha−1 every 8 to 10 days. Disease severity (percentage of EB) represents the mean value of four independent repetitions
Assay development for real-time PCR
Primers designed (Schuhegger et al. 2006) were highly specific for the quantification of A. alternata. Here, cross-reactions were only found for A. tenuissima. The sequences used for the primer design of A. solani were species-specific without any cross-amplification between the two species. However, the primer pair used to quantify A. solani yielded some weak positive signals with a few other non-related fungi, probably due to very close identity of primer and non-target DNA sequences of these fungi (Table 1). Among them were species like Pyrenochaeta lycopersici, Stemphylium sarciniforme or Thielaviopsis basicola which are not pathogenic to potato. PCR products of Colletotrichum coccodes, Fusarium solani or Phytophthora infestans, which may also cause leaf injury on potato, only yielded weak signals.
Real-time PCR
In addition to visual observations, real-time PCR analysis was carried out to determine the amount of Alternaria DNA in potato leaves. In 2003 and 2004, leaves from the mid-canopy were sampled and further evaluated for the amount of Alternaria DNA.
The real-time PCR analysis detected the presence of Alternaria DNA in almost all samples at quite early stages of plant development. Analysis revealed that A. alternata DNA was already observed at the first sampling dates of both years (June 16th, 2003 and June 7th, 2004). In 2004, A. solani was similarly detected on June 7th, whereas in 2003 it was found significantly later on Aug 5th. The use of species specific primers enabled the monitoring of fungal spread of A. solani and A. alternata within the leaves. Annual differences were observed for the development of Alternaria species. Differences in the increase of fungal DNA were observed in 2003 and 2004 where A. alternata was predominant from the beginning of sampling, albeit at very low DNA levels. In 2003, a steady increase of DNA levels yielded 230 pg A. alternata DNA per mg fresh weight (FW). In contrast, A. solani was detected only on the last two sampling dates. Comparing the amounts of DNA on August 18th, it was obvious that DNA levels of A. alternata were almost twice as high as those of A. solani (Fig. 3).

Progression of A. solani and A. alternata growth measured by real-time PCR in 2003 and 2004. Real-time PCR analysis was carried out for leaves sampled from the middle leaf section of the potato plant. Vertical bars indicate standard deviation. Columns with the same letter are not significantly different (Tukey’s b test, P = 0.05)
In 2004, A. solani and A. alternata developed in parallel beginning from June 7th onwards. DNA of both species was found to be present at almost equal levels throughout the season. However, total amounts of DNA were lower than in 2003.
From 2005 onwards, A. solani was found to be the predominant species, with high quantities of fungal DNA. Although small amounts of DNA of both species were detected at the beginning of the season, significantly higher amounts of A. solani DNA could be detected as the season progressed. In contrast, in 2005 and 2007 low A. alternata DNA values were observed which remained at low levels until the end of the season. Highest values of A. alternata DNA were approx. 3 pg DNA per mg FW, whereas levels of A. solani were much higher on each sampling date and reached 20–60 pg per mg FW at the end of the season (Fig. 4a).

Progression of A. solani and A. alternata growth measured by real-time PCR in 2005 (4a) and 2007 (4b). In order to follow up species development in more detail, potato plants were divided into three leaf layers (bottom, middle and upper leaf layer). Vertical bars indicate standard deviation. Columns with the same letter are not significantly different (Tukey’s b test, P = 0.05)
In 2005 and 2007, leaf samples were taken simultaneously from three different plant sections. Although symptoms were only observed on lower leaves, analysis of the middle and top third of the canopy revealed Alternaria DNA prior to the development of EB symptoms. Therefore, real-time PCR allows the detection of fungal growth within different plant sections. The largest increase in DNA amounts took place in the middle leaf section in 2005 (Fig. 4a). Higher amounts of A. solani DNA were detected in 2007 in the upper leaf layers (Fig. 4b). Here, the largest increase of fungal DNA amounts took place during a disease epidemic with severe symptom increase.
To understand the relationship between the amount of Alternaria DNA measured by real-time PCR and the development of leaf necroses, a linear regression was calculated (Fig. 5). In the beginning, low DNA levels were observed similar to low disease scorings. However, a stronger increase in DNA was observed almost without exception for A. solani. Here, DNA level increased proportionally with an increase of leaf necrotic surface, whereas those of A. alternata remained at a low level. Real-time PCR results and percentages of leaf necrotic area obtained for A. solani were closely related (r = 0.71), whereas no causal relationship was found for A. alternata (r = 0.12). The regression was carried out for 60 individual samples in 2005.

Relationship between visual disease assessments (percent leaf area infected) and DNA levels of A. solani (A) and A. alternata (B) measured by real-time PCR. Regression was carried out for 60 individual samples taken throughout 2005 from the location Straubing
Detection of A. solani and A. alternata after fungicide treatment
Comparing the EB severity of non-treated plants with fungicide treated variants, amounts of fungal DNA were differently reduced. Fungicidal treatments with either azoxystrobin or mancozeb resulted in a significant decrease in the amounts of fungal DNA, which was quite pronounced for leaves of the middle and upper leaf canopy taken at the end of the season (sampling date 2005: Aug 12th, 2006: Sept. 1st) (Fig. 6a,b). Statistical analysis revealed significant differences in DNA levels of A. solani with respect to the specific fungicide treatment. In contrast, differences in the DNA amount of A. alternata were only marginal. Highest DNA levels were found for A. solani in the EB non-treated control in both years and all leaf sections. In 2005, 56.6 pg DNA/mg FW was determined for A. solani in the middle leaf layers, whereas 7.5 pg DNA/mg FW was found in the upper leaf sections (Fig. 6a). Real-time PCR revealed that the application of azoxystrobin resulted in a significant decrease in A. solani DNA levels as compared to mancozeb. While 0.6 pg DNA/mg FW (middle leaf section) and 0.2 pg DNA/mg FW (upper leaf section) of A. solani were determined in the azoxystrobin-treated samples in 2005, mancozeb treated plots resulted in a less pronounced reduction of DNA levels. Here, analysis yielded 8.0 pg DNA/mg FW (middle leaf section) and 2.2 pg DNA/mg FW (upper leaf section). Comparable results were observed for the consecutive year 2006 (upper leaf layer) when a significant reduction was found for A. solani in azoxystrobin (3.68 pg DNA/mg FW) and mancozeb (9.05 pg DNA/mg FW) treated plots compared to the non-treated plot (48.04 pg DNA/mg FW) (Fig. 6b).

DNA content of A. solani and A. alternata depending on the applied fungicides measured by real-time PCR in 2005 (6a) and 2006 (6b). In order to prevent late blight, all plots (including the EB non-treated plot) were treated with the fungicide Ranman (cyazofamid at 400 g l−1) as a cover spray at a dose of 0.2 l ha−1 every 8 to 10 days. Ranman has no efficacy against EB. Fungicide plots were treated at weekly intervals with either azoxystrobin (125 g a.i. ha−1) or mancozeb (1350 g a.i. ha−1). Vertical bars indicate standard deviation. Columns with the same letter are not significantly different (Tukey’s b test, P = 0.05)
In contrast to A. solani, low quantities of A. alternata DNA were found in the EB non-treated plots in 2005 and 2006. Here 0.42 and 1.01 pg DNA/mg FW, respectively, was found for leaf samples from the upper leaf section. With respect to the different fungicide treatments, reduction of DNA levels was statistically significant (upper leaf section).
Discussion
In this study, a real-time PCR assay was developed to detect and quantify A. solani and A. alternata DNA in potato leaves. The results demonstrate that the developed real-time PCR protocol is a sensitive and reproducible method for in planta quantification of Alternaria spp. during potato leaf colonization. The gradual upward progression, as seen by visual disease ratings, was similarly reflected by real-time PCR according to the development of the fungal DNA content.
As described above, visual assessment of symptoms does not guarantee a distinct differentiation of pathogens, as both species could be isolated simultaneously from the same lesions. Therefore, reliable and specific methods are necessary to improve the diagnostics of both Alternaria species. Additionally, sensitive and quantitative detection techniques would be helpful to characterize dynamics of Alternaria on potato.
Differences in genome sequences between species enabled the development of species-specific primers. According to Guillemette et al. (2004), the ITS region of nuclear rDNA is the main genomic region targeted for PCR primer development. The ITS primers used in this study proved to be highly specific for A. solani and A. alternata. Weak positive amplification signals in the case of A. solani primers by unrelated fungi were found in a few cases (Table 1). These amplicons were obtained from pure fungal DNA. However, such a scenario is very unlikely to occur in nature. They indicate sufficient sequence similarity – particularly at the 3’ends of the primers - to DNA sequences of the non-target fungi, allowing incomplete annealing and subsequent poor amplification. This problem could probably be solved by increasing stringency, e.g. by increasing the annealing temperature. Since cross-reactions were mainly found for species non-pathogenic to potato and at unrealistically high DNA concentrations, they were not considered a drawback for this study. According to (Pyror and Michailides 2002) A. alternata and A. tenuissima group as a single section as indicated by phylogenetic analysis. In contrast to morphological and cultural diagnostics which take about 4 to 5 days, the test has the advantage that it is less laborious and can be completed within one working day from DNA extraction to PCR detection. It also represents a more objective way to monitor fungal growth than phenotypic disease rating scores.
According to the dilution series of Alternaria DNA, both species can be accurately quantified in the absence or presence of host DNA. In contrast to the sensitivity of conventional PCR assays, which is not sufficient to detect early infection stages at low disease incidence (Schena et al. 2004), the real-time PCR method described here was much more sensitive in detecting early or latent infections. Detection thresholds were as low as 100 fg, enabling the application of real-time PCR in field experiments.
As DNA analysis was impaired at higher leaf necroses, a linear regression could only be calculated up to 50 % leaf necrotic surface. Similarly, in the pathosystem M. graminicola on wheat, high correlations were only observed up to 40 % leaf necrosis (Selim et al. 2013). Nevertheless, PCR analysis of leaf samples taken at various times allowed monitoring of the growth progression of Alternaria species. Although symptom appearance was very inconspicuous, real-time PCR has proven to be very effective for the quantification of Alternaria DNA levels at very early or even latent stages of infections, which would help to improve diagnosis. Concerning latent infections, real-time PCR detected fungal DNA in the leaf although no characteristic blight symptoms were observed during sampling. Such detection of latent infections can support the prediction of an epidemic on leaves located higher up on the plant.
Results from 2005 and 2007 showed that as the season progressed, leaves from different canopy sections obviously expressed different fungal DNA levels, confirming that leaf age may influence Alternaria spp. development. According to Rotem (1994), symptom development is associated with a shift of plant metabolism. Nutrients are more readily available to the pathogen in older leaves than in younger leaves because they are no longer immediately processed and incorporated. This would allow the pathogen to colonize older leaves more rapidly and extensively than younger leaves (Wharton and Kirk 2007). These findings were supported by the identification of higher amounts of Alternaria DNA in older leaves at first sampling dates. With increasing leaf age, successively higher located leaves were affected by EB, reflected by higher DNA levels. As epidemics mainly intensify at flowering and reach their maximum intensity in senescent plants, Alternaria diseases have been considered as diseases of aging plants (Rotem 1994). In addition, rapid increases in EB disease towards the end of the growing season were also thought to be partly due to the reduced resistance of the foliage (Horsfield et al. 2010). Real-time PCR analyses confirmed these results, as higher leaf sections were increasingly affected as the season progressed and leaves aged.
Analysis revealed significantly different amounts of Alternaria DNA of both pathogens and in all years from 2003 to 2007. A. solani was predominantly found in all years of investigation, except for 2003. In 2003 only, A. alternata was found at higher DNA levels. Viskonti and Chelkowski (1992) stated that species development is strongly influenced by weather conditions. Despite the lack of evidence, it is more likely that prevailing environmental conditions have favoured A. alternata development in 2003. In the following year, 2004, a more equal distribution of both species was revealed by real-time PCR. In fact, A. solani was found at higher DNA amounts throughout the season at Straubing in the years investigated. Even though both species were found in almost all years, more consistent and higher DNA levels were expressed by real-time PCR analysis for A. solani rather than for A. alternata. According to previous research, EB is mainly caused by A. solani (Viskonti and Chelkowski 1992). Rotem (1994) described A. solani as being highly pathogenic to potato, penetrating potato tissue directly within 4 to 6 h. Higher DNA values of A. solani can be explained by its higher aggressiveness compared to A. alternata. Artificial inoculations of potato plants in the greenhouse with A. alternata revealed no or only little symptoms (at least 1 % disease severity) whereas inoculations of A. solani resulted in high disease values (Leiminger, data unpublished, Stammler et al. 2014). According to previous results A. alternata has been stated as a saprophyte which may penetrate leaves only as a secondary invader (Rotem 1994).
Up to now, the application of foliar fungicides is the most commonly used method used for Alternaria disease control. Fungicides with different modes of action were shown to be differentially effective (Horsfield et al. 2010). Real-time PCR has been proven as a diagnostic assay to investigate the impact on pathogen development after fungicide application. Although fungicide treatments resulted in a significant DNA reduction compared to the non-treated control, the efficiency of mancozeb and azoxystrobin were quite different. Results showed that azoxystrobin led to significantly lower DNA levels of A. solani than mancozeb. These results are in agreement with those reported by Rosenzweig et al. (2008), showing that azoxystrobin gave the best early blight control, provided that no shift in sensitivity had occurred. According to Pasche et al. (2004), azoxystrobin turns out to be highly effective whilst mancozeb is known as only moderately effective in the control of A. solani (Stevenson and James 2007). In contrast to A. solani, the fungicidal activity on A. alternata was very low. Here, no differences in the reduction of A. alternata were observed between fungicide treated and non-treated plots. However A. alternata DNA levels found in the Alternaria non-treated control were very low. Accordingly, perennial real-time PCR results showed that leaf necrosis were caused invariably by A. solani at Straubing. Real-time PCR therefore presents an accurate, specific and quantitative method for determining the role of different inocula sources. Quantitative methods could be used for dynamic population studies throughout the growing season. Deeper insights into plant-pathogen interactions will support future disease control through better timing and choice of fungicide applications.
The etection of Alternaria species in plant material became more difficult at high infection levels. At the very last sampling dates, real-time PCR did not yield any results, although leaves were heavily infected. This is probably due to the lack of live mycelia. The reduction in fungal DNA at the latter stages of the disease epidemic suggests that part of the fungal hyphae have died off, or that PCR inhibitors that are present at much higher levels in diseased leaf tissues than in those without symptoms, have increased (Singh et al. 1998). Fungal mycelia in plant tissues deteriorate upon sporulation. Therefore PCR detection might not be possible after tissues have become totally necrotic and DNA cannot easily be extracted from spores by conventional techniques, which is a weakness of the method used.
If PCR-based detection is still desirable in such cases, methods capable of extracting DNA from mature spores should be used, since spore production is favoured with increasing leaf necrosis (Pasche et al. 2005).
In conclusion, we have developed a rapid, sensitive and reliable real-time PCR assay based on the amplification of ITS rRNA for the detection and quantification of viable Alternaria spp. in potato. This technique now can be used for screening of materials for the presence of Alternaria pathogens, which would further help to understand the dynamics of Alternaria spp. in field grown potato. The qPCR assay reported here may be used to provide a means of estimating colonization of both Alternaria species and opens up the opportunity to study the progression of A. solani and A. alternata within the host plant.
References
Bahnweg, G., Schulze, S., Möller, E. M., Rosenbrock, H., Langebartels, C., & Sandermann, H. (1998). DNA isolation from recalcitrant materials such as tree roots, bark, and forest soil for the detection of fungal pathogens by polymerase chain reaction. Analytical Biochemistry, 262, 79–82.
Bäßler, E., Asensio, N., Leiminger, J., Hausladen, H., Bahnweg, G., & Zinkernagel, V. (2004). Untersuchungen zum Auftreten und zur Bekämpfung von Alternaria solani und Alternaria alternata an Kartoffeln, Investigations on the appearance and control of Alternaria solani and Alternaria alternata on potato. Mitt. Biol. Bundesanst Land—Forstwirtschaft, 396, 100–101.
Boiteux, I. S., & Reifschneider, F. J. B. (1989). Potato early blight caused by Alternaria alternata in Brazil. Fitopatologia Brasileira, 14, 117.
Granovsky, A. A., & Peterson, A. G. (1954). Evaluation of potato injury caused by leafhoppers, flea beetles, and early blight. Journal of Economic Entomology, 47, 894–902.
Guillemette, T., Iacomi, B., & Simoneau, P. (2004). Conventional and real-time PCR based assay for detecting pathogenic Alternaria brassicae in cruciferous seed. Plant Disease, 88, 490–496.
Hausladen, H., & Leiminger J. (2007). Potato early blight in Germany (Alternaria solani – Alternaria alternata), PPO-Special report no. 12, Westerdijk, C.E., Schepers, H.T.A.M., (HRSG), Applied Plant Research BV, Wageningen.
Horsfield, A., Wicks, T., Davies, K., Wilson, D., & Paton, S. (2010). Effect of fungicide use strategies on the control of early blight (Alternaria solani) and potato yield. Australasian Plant Pathology, 39, 368–375.
Konstantinova, P., Bonants, P., van Gent-Pelzer, M., van der Zouwen, P., & van den Bulk, R. (2002). Development of specific primers for detection and identification of Alternaria spp. in carrot material by PCR and comparison with blotter and plating assays. Mycological Research, 106, 23–33.
Latorse, M. P., Schmitt, F., Peyrard, S., Veloso, S., & Beffa, R. (2010). Molecular analysis of Alternaria populations early blight causal agents in potato plants. In C. E. Westerdijk & H. T. A. M. Schepers (Eds.), PPO special report 14 (pp. 179–186). Wageningen: Applied Plant Research BV.
Leiminger, J. H., & Hausladen, H. (2012). Early blight control in potato using disease-orientated threshold values. Plant Disease, 96, 124–130.
Pasche, J. S., Wharam, C. M., & Gudmestad, N. C. (2004). Shift in sensitivity of Alternaria solani in responses to QoI fungicides. Plant Disease, 88, 181–187.
Pasche, J. S., Piche, L. M., & Gudmestad, N. C. (2005). Effect of the F129L mutation in Alternaria solani on fungicides affecting mitochondrial respiration. Plant Disease, 89, 269–278.
Pavón, M. Á., González, I., Pegels, N., Martín, R., & García, T. (2010). PCR detection and identification of Alternaria species-groups in processed foods based on the genetic marker Alt a 1. Food Control, 21, 1745–1756.
Pavón, M. Á., González, I., Martín, R., & García, T. (2012). A real-time reverse-transcriptase PCR technique for detection and quantification of viable Alternaria spp. in foodstuffs. Food Control, 28, 286–294.
Pyror, B. M., & Michailides, T. J. (2002). Morphological, pathogenic, and molecular characterization of Alternaria isolates associated with Alternaria late blight of pistachio. Phytopathology, 92, 406–416.
Rosenzweig, N., Atallah, Z. K., & Stevenson, W. R. (2008). Evaluation of QoI fungicide application strategies for managing fungicide resistance and potato early blight epidemics in Wisconsin. Plant Disease, 92, 561–568.
Rotem, J. (1994). The genus Alternaria, biology and pathogenicity. Minnesota: APS Press.
Schena, L., Nigro, F., Ippolito, A., & Gallitelli, D. (2004). Real-time quantitative PCR: a new technology to detect and study phytopathogenic and antagonistic fungi. European Journal of Plant Pathology, 110, 893–908.
Schuhegger, R., Ihring, A., Gantner, S., Bahnweg, G., Knappe, C., Vogg, G., Hutzler, P., Schmid, M., van Breusegem, F., Eberl, L., Hartmann, A., & Langebartels, C. (2006). Induction of systemic resistance in tomato by N-acyl-L-homoserine lactone-producing rhizosphere bacteria. Plant, Cell and Environment, 29, 909–918.
Selim, S., Roisin-Fichter, C., Andry, J. B., Bogdanow, B., & Sambou, R. (2013). Real-time PCR to study the effect of timing and persistence of fungicide application and wheat varietal resistance on Mycosphaerella graminicola and its sterol 14α-demethylation-inhibitor-resistant genotypes. Pest Management Science. doi:10.1002/ps.3525.
Singh, R. P., Singh, M., & King, R. R. (1998). Use of citric acid for neutralizing polymerase chain reaction inhibition by chlorogenic acid in potato extracts. Journal of Virological Methods, 74, 231–235.
Stammler, G., Böhme, F., Philippi, J., Miessner, S., & Tegge, V. (2014). Pathogenicity of Alternaria-species on potatoes and tomatoes. In C. E. Westerdijk & H. T. A. M. Schepers (Eds.), PPO special report 16 (pp. 85–96). Wageningen: Applied Plant Research BV.
Stevenson, W. R., & James, R. V. (2007). Evaluation of fungicides to control early blight of potato – Hancock. Plant Disease Management Reports, 2, V050.
Tylor, E., Bates, J., Kenyon, D., Maccaferri, M., & Thomas, J. (2001). Modern molecular methods for characterization and diagnosis of seed-borne fungal pathogens. Journal of Phytopathology, 83, 75–81.
Udayashankar, A. C., Chandra Nayaka, S., Archana, B., Anjana, G., Niranjana, S. R., Mortensen, C. N., Lund, O. S., & Prakash, H. S. (2012). Specific PCR-based detection of Alternaria helianthi: the cause of blight and leaf spot in sunflower. Archives of Microbiology, 194, 923–932.
van der Waals, J., Korsten, L., & Denner, F. D. N. (2003). Early blight in South Africa: knowledge, attitudes and control practices of potato growers. Potato Research, 46, 27–37.
Viskonti, A., & Chelkowski, J. (1992). Alternaria Biology, plant diseases and metabolites, eds. Amsterdam: Elsevier Verlag.
Wharton, P. S., & Kirk, W. W. (2007). Early blight michigan potato diseases series. MSU Extension bulletin, E-2991.
Zur, G., Shimoni, E. M., Hallerman, E., & Kashi, Y. (2002). Detection of Alternaria fungal contamination in cereal grains by a polymerase chain reaction-based assay. Journal of Food Protection, 65, 1433–1440.
Acknowledgements
We thank Ralph Hückelhoven for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Leiminger, J., Bäßler, E., Knappe, C. et al. Quantification of disease progression of Alternaria spp. on potato using real-time PCR. Eur J Plant Pathol 141, 295–309 (2015). https://doi.org/10.1007/s10658-014-0542-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10658-014-0542-2