
Summary
Neratinib, an irreversible pan-HER tyrosine kinase inhibitor, has been approved for the treatment of HER2-positive (HER2+) early-stage and brain metastatic breast cancer. Thus far, the pharmacology effects and pharmacodynamics of neratinib have been well studied. However, the disposition of neratinib and its influencing factors in vivo remain unclear. P-glycoprotein (P-gp), one of the most extensively studied transporters, substantially restricts penetration of drugs into the body or deeper compartments (i.e., blood-brain barrier, BBB), regarding drug resistance and drug-drug interactions. Thereby, the aim of this study was to investigate the influence of verapamil (a P-gp inhibitor) on the pharmacokinetics of neratinib in rats. Here, we have established a high specific, selective and sensitive ultra-performance liquid chromatography-tandem mass spectrometric (UPLC-MS/MS) method to quantify plasma concentrations of neratinib in rats. Pharmacokinetic results showed that verapamil significantly increased the system exposure of neratinib, as Cmax increased by 2.09-fold and AUC0-t increased by 1.64-fold, respectively. Additionally, the in vitro transport of neratinib was evaluated using Madin-Darby canine kidney II (MDCK II) and human MDR1 gene overexpressed MDCK (MDCK-MDR1) cell line models. As a result, the net flux ratio was over than 2 and decreased over 50% by verapamil, suggesting that neratinib was a substrate of P-gp. Hence, our findings have highlighted the important role of P-gp in the system exposure of neratinib in vivo, and drug-drug interaction should be considered when coadministration of P-gp inhibitors with neratinib. These findings may support the further clinical development and application of neratinib.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neratinib (HKI-272) is an oral, irreversible pan-inhibitor of human epidermal receptor (HER) tyrosine kinase inhibitor, including HER1 (also known as EGFR), HER2 and HER4 [1, 2]. The drug reduces phosphorylation and blocks downstream signaling of these receptors by covalently binding to the conserved cysteine residues in receptors [3]. Neratinib is the first intensive adjunctive therapy approved by the US FDA in 2017 for HER2-positive (HER2+) early-stage breast cancer patients who have completed standard trastuzumab-based chemotherapy and have not progressed but still with high risk [1, 2]. When in combination with capacitation, neratinib has been approved for the treatment of advanced or metastatic HER2+ breast cancer who have received two or more anti-HER2 agents [4, 5]. A phase III study of neratinib has demonstrated significant improvement in 5-year disease-free survival rate [6]. Furthermore, the drug, alone or in combination, has shown antitumor efficacy for other solid tumors (such as non-small cell lung cancer, colorectal cancer and glioblastoma) [1, 7, 8]. Thus far, the pharmacology effects and pharmacodynamics of neratinib have been well studied. However, the disposition of neratinib and its influencing factors remain unclear.
The ATP binding cassette (ABC) membrane transporter P-glycoprotein (P-gp), formerly known as multidrug resistance protein 1 (MDR1), is a transmembrane efflux pump, which can employ the energy generated by ATP hydrolysis to excrete a large number of endogenous and exogenous substrates out of the cell [9]. P-gp is widely distributed in multiple organs, such as intestine, liver, kidney, blood-brain barrier (BBB), placenta and lymphocytes [9]. Due to its diverse tissue distribution, P-gp-mediated efflux can be physiologically considered as a defense mechanism against the penetration of xenobiotics into the body or deeper compartments [9, 10]. However, from the therapeutic perspective, P-gp functions to 1) limit absorption from the intestine to the systemic circulation with orally administered drugs, 2) prevent drugs from crossing blood-brain barrier into the central nervous system, and 3) contribute to the active excretion of drugs from liver and kidney tissue [10, 11]. Therefore, the inhibition or induction of P-gp may increase or decrease the systemic exposure of its substrates, which is related to drug resistance and drug-drug interaction.
Clinically, approximately 24-28% of HER2+ breast cancer patients will develop brain metastases, which results in poor prognosis [12]. Trastuzumab is the first-line drug for HER2+ breast cancer. However, its ability to cross the BBB is limited due to the high molecular weight (145,531 Da) [13]. Therefore, small-molecular tyrosine kinase inhibitors, such as neratinib (with a lower molecular weight, 557 Da), are promising for the treatment of breast cancer patients with BM as its theoretical ability to cross the BBB [14]. In fact, neratinib has limited activity in the CNS when given alone [15, 16]. In addition, a clinical trial reported negligible neratinib concentrations in the cerebrospinal fluid (CSF) in a limited number of specimens from patients with progressive HER2+ breast cancer brain metastases who received neratinib monotherapy and undetectable neratinib concentrations in parenchymal CNS samples [16]. What’s more, the factors influencing the oral bioavailability and the ability to cross the BBB of neratinib have not fully established. As P-gp is one of the most investigated efflux transporters and present in many key organs in drug disposition such as small intestine, BBB, kidney, and liver. Therefore, it is necessary to determine the role of P-gp in the bioavailability of orally administered neratinib.
In this study, a UPLC-MS/MS method for the determination of neratinib in rat plasma was established and validated. Then the effect of verapamil (a known P-gp inhibitor) on the pharmacokinetics of neratinib in rats was determined using this sensitive and reliable UPLC-MS/MS method. Furthermore, Madin-Darby canine kidney (MDCK) II cells and MDR1 overexpressed MDCK (MDCK-MDR1) cells were used to identify whether neratinib was a substrate of P-gp. We provided the first evidence that P-gp is a major determinant of the bioavailability of orally administered neratinib.
Materials and methods
Chemicals and reagents
Neratinib (purity > 99%) and Verapamil (purity > 99%) was purchased from MedChemErpress (Monmmouth Junction, NJ, United States). Dimethyl sulphoxide (DMSO) and Sodium chloride solution (0.9%) were analytical grade and purchased from Sigma-Aldrich (St Louis, MO, United States). Acetonitrile, methanol and t-butyl methyl ether, which were LC–MS grade, were bought from Merk KGaA (Darmstadt, Germany).
Instruments and conditions
The analysis was performed using Waters ACQUITY UPLC system (Milford, MA) with a Hypersil GOLD™ column (100 × 2.1 mm, 1.9 μm particle size; Thermo Scientific). The temperature of the column was set at 40 °C. The samples were separated using a gradient of water (containing 2.5 mM ammonium acetate and 0.1% formic acid, mobile phase A) and acetonitrile (containing 0.1% formic acid, mobile phase B) at a flow rate of 0.35 mL/min. The gradient elution programs were as follows: 30% B at 0-0.2 min, 30-35% B at 0.2-1.2 min, 35-95% B at 1.2-1.5 min, 95% B at 1.5-2.0 min, 95-30% B at 2.0-3.0 min, and 30% B at 3.0-4.0 min. The injection volume was 5 μL, and the auto-sampler chamber temperature was set at 10 °C.
Mass spectrometry analysis was performed on the Xevo TQD MS (Waters) using the electrospray ionization source (ESI) in positive ionization mode. The selected monitoring conditions were as follows: capillary voltage, 4.0 kV; cone voltage, 50 V; source temperature, 150 °C; desolvation temperature, 450 °C; desolvation gas flow, 900 L/h; cone gas flow, 50 L/h. The precursor to daughter transitions of neratinib and sorafenib (IS) was firstly monitored, and the optimal collision energy for neratinib and IS was 30 and 35 V, respectively. Then quantification was conducted using MRM with the precursor-to-product ion transitions at m/z 557.50 > 111.98 for neratinib and m/z 465.31 > 252.11 for IS, respectively.
Preparation of plasma samples
Standard stock solutions of neratinib and IS (sorafenib) were all prepared in DMSO at the concentration of 10 and 2 mg/mL, respectively. When used, stock solutions were diluted with mixed solvent of DMSO and methanol (1:4, v/v) to the desired concentrations as working solutions. 100 μL aliquot of rat plasma (blank or containing neratinib) was mixed with 1.5 mL of methyl tert-butyl ether containing IS (sorafenib, 150 ng/mL) and extracted by vortex for 2 min. After centrifugation at 5000 g for 15 min (4 °C), the upper organic portion was transferred to a clean tube and dried using SP Genevac Ltd.'s miVac Centrifugal Vacuum Concentrator (Warminster, England). Finally, the residue was reconstituted to 150 µL using acetonitrile/water (50:50, v/v). After centrifugation (12,000 g, 15 min), the supernatant was subjected to UPLC-TQD/MS (Waters) for analyses.
Method validation
Selectivity
The purpose of selectivity investigation is to evaluate whether there is obvious interference of other substances at the peak position of analytes or IS in matrix. Blank rat plasma containing neratinib and IS was prepared as described in Preparation of plasma samples section and analyzed by UPLC-TQD/MS, and blank matrix was used as control.
Linearity and lower limit of quantification (LLOQ)
Neratinib was added into blank rat plasma at different concentrations (20-4000 ng/mL) to be prepared as standard curve samples. The internal standard method was used for calibration. The linear regression equation was obtained by plotting the peak area ratio of the analyte (neratinib) to IS versus six-point concentrations (20-4000 ng/mL) of the analyte in plasma. The LLOQ was obtained by the signal-to-noise ratio of 10:1.
Accuracy and precision
Six replicates of quality control (QC) samples at low (40 ng/mL), media (400 ng/mL), and high (4000 ng/mL) concentrations in rat plasma were prepared as described in Preparation of plasma samples section. Then the inter-day and intra-day accuracies and precisions were determined. Accuracy was expressed as % recovery according to Eq. (1). Precision was expressed by coefficient of variation (CV), which was calculated according to Eq. (2). The maximum allowed deviation of CV% was within 15%.
Extraction recoveries and matrix effects
Three different concentrations of neratinib QC samples (40, 400 and 4000 ng/mL) in rat plasma were prepared and named as “spiked matrix”. The peak areas of QC samples detected by UPLC-TQD/MS were described as “A”. Blank rat plasma matrix was prepared and dried as described in Preparation of plasma samples section. Then, the residue was reconstituted to 150 µL of acetonitrile/water (50:50, v/v) containing neratinib and IS, and make the concentration of neratinib and IS the same as QC samples. The peak areas of these samples detected by UPLC-TQD/MS were named as “B”. For “neat solution” samples, neratinib and IS were directly added into 150 µL of acetonitrile/water (50:50, v/v) at the same concentration as QC samples. The peak areas of “neat solution” samples detected by UPLC-TQD/MS were named as “C”. The extraction recoveries and matrix effects were calculated according to Eq. (3) and (4), respectively.
Stability
The stabilities of neratinib at 40, 400, and 4000 ng/mL in different storage conditions were evaluated, including long-term (-80 °C for 10 days) and three freeze–thaw cycle stabilities of neratinib in plasma, short term stability (ambient temperature for 4 h and 4 °C for 24 h) in 50% acetonitrile solution. Evaluation was carried out in triplicate to quantify the %recovery of neratinib concentrations after different operations to the concentration at time zero.
Pharmacokinetic studies
Animals
Male Sprague–Dawley rats weighing 200–220 g, purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China), were used for pharmacokinetic (PK) studies. All animals were raised in standardized environmental conditions (12 h light/dark schedule), rearing standard diet and water, and allowed to adapt to the environment for one week before inclusion in the experiment. All procedures were adhered to the National Institutes of Health guide for the care and use of Laboratory Animals (NIH Publications No. 8023, revised 1978) and were granted by the Medical and Research Ethics Committee of College of Medicine, Henan University.
Experimental design
Before PK studies, a cannula was inserted into the jugular vein of the anesthetized rat by 10% chloral hydrate and administrated carprofen subcutaneously after the surgery. To evaluate the effects of verapamil on the pharmacokinetic of neratinib, the rats were randomly divided into two groups of six rats each, the control and treatment group. The animals were fasted overnight prior to drug administration but water was provided ad libitum. Neratinib and verapamil were prepared in a suspension using normal saline containing 0.5% sodium carboxymethyl cellulose (CMC-Na, the suspending agent). The treatment group of rats were pretreated with verapamil at a dose of 10 mg/kg (oral gavage), and the control group of rats were only administered with equal amounts of vehicle. After 0.5 h, neratinib was orally administered to rats by gavage at a dose of 40 mg/kg. Approximately 200 μL blood samples were collected from jugular vein at 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 24 and 48 h after administration of neratinib. The rats were then immediately injected with the same amount of saline through a jugular vein cannula at each sampling time point. Blood samples were immediately centrifuged at 4 °C (8000 g,10 min) and the plasma was collected, which was stored at -80 °C until analysis.
Samples preparation
100 µL of plasma was transferred to a new tube, samples were treated with protein precipitation assay according to the Preparation of plasma samples section. A calibration curve was prepared to quantify the amount of neratinib in plasma samples.
Pharmacokinetics analysis
The non-compartmental approach was used to analyze the plasma data after oral administration. The pharmacokinetics parameters estimation, including the area under the plasma concentration–time curve (AUC), maximal plasma concentration (Cmax), the time for the maximal plasma concentration (Tmax), half time (t1/2) and clearance (CL), were performed using WinNonlin software (Pharmsight, Mountain View, CA).
Cell culture
Madin Darby canine kidney (MDCK II) cell line was obtained from the Cell Bank of the Chinese Academy of Science (Shanghai, China) and were routinely cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum (HyClone, UT, USA) and 100 IU/mL penicillin/streptomycin. Cells were cultured at 37 °C in a humidified atmosphere with 5% CO2. Multidrug resistance 1 gene (MDR1) overexpressed MDCK II cells (MDCK-MDR1) were constructed in our previous publications. The culture conditions of MDCK-MDR1 cells were the same as those of MDCK cells.
Neratinib cellular uptake assay
In this experiment, MDCK II cells or MDCK-MDR1 cells were planted in 6-well plate at the destiny of 2 × 105 cells/well for 48 h and used to evaluate the uptake of neratinib. After discarding the old culture medium, the cells were washed twice with 2 mL of 37 °C prewarmed Hanks’ balanced salt solution (HBSS) buffer (pH = 7.2). Then cells were incubated at 37 °C in 2 mL HBSS buffer containing neratinib at the final concentrations of 1.25 and 5 µM. P-gp inhibitor (verapamil, 100 µM), when used, was co-incubated with neratinib. After incubation for 2 h, the incubation media was removed, and cells were washed twice using pre-cooled HBSS buffer. Then cells were collected into 400 µL of ice-cold MeOH:H2O (1:1, v/v) and sonicated in an ice bath for 15 min. After centrifugation at 12,000 g (4 °C) for 10 min, the supernatant was collected and injected into UPLC-TQD/MS to determine intracellular amount of neratinib.
Bidirectional transport experiments
MDCK II and MDCK-MDR1 cells were seeded into 12-well Transwell polycarbonate membrane inserts (12 mm diameter inserts, 0.4 μm pore size, Corning Costar) at a density of 6 × 105 cells/well in complete DMEM medium. The transepithelial electrical resistance (TEER) of the polarized cell monolayer was monitored until the tight confluent cell monolayer formed with the TEER values over 200 Ω∙cm2. Transport studies of neratinib were performed from the apical (AP)-to-basolateral (BL) (AP-BL) and BL to AP (BL-AP) directions. All the bidirectional transport experiments were conducted in HBSS buffer (pH = 7.2) with gentle shaking (60 rpm) at 37 °C. On the day of the experiment, cells were washed twice with warm (37 °C) HBSS and preincubated at 37 °C for 30 min.
Briefly, neratinib that dissolved in HBSS at a final concentration of 5 μM was added to either apical or basolateral compartment as donor compartment. For receiver compartment, blank HBSS was added. The effects of verapamil (100 μM) on the transport of neratinib were investigated by adding to both compartments during the incubation period. The volume of incubation medium on the apical side (AP) and basolateral side (BL) were 0.5 mL and 1.5 mL, respectively. Samples (100 μL) were withdrawn at specific time points (0.5, 1, 1.5 and 2 h) from the receiver compartment and then replaced with 100 μL of blank HBSS at 37 °C. For analysis after incubation, all samples were mixed with 50 μL acetonitrile, and after centrifugation at 4 °C (12,000 g, 10 min), the supernatant was subjected into UPLC-TQD/MS system.
The apparent permeability coefficient (Papp) for both directions, including from the AP side to the BL side (Papp,AP-BL) and from the BL side to the AP side (Papp,BL-AP), was calculated according to the following Eq. (5).
where Papp is the apparent permeability coefficient (cm/s), dQ/dt is the rate at which neratinib permeates in the receiver chamber (nmol/s), A is the area of polycarbonate membrane (A = 1.12 cm2), and C0 is the initial concentration of neratinib in the donor compartment (μM).
Further, the efflux ratio (ER) was calculated according to Eq. (6). The net flux ratios were calculated by dividing the ER obtained in MDCK-MDR1 cells by the ER obtained with the parent line MDCK II. A net flux ratio over 2 indicated the efflux of test compound was specifically mediated by P-gp [17].
Immunoblot analysis
Protein expression of P-gp in MDCK II and MDCK-MDR1 cells was determined by immunoblotting. In brief, protein samples (30 µg) were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (10% resolving and 5% stacking gels) and then transferred to a polyvinylidene fluoride (PVDF) membrane. After blocked for 2 h in 5% nonfat milk, membrane was incubated with primary antibody (1:1000 dilution for P-gp and β-actin antibody) overnight at 4 °C, and then incubated with HRP-conjugated secondary antibody for 2 h at room temperature. A chemiluminescence-enhanced image of the target protein bands was detected using autoradiography. Grayscale scan and semi-quantitative analysis of band intensities was performed using the Quantity One software.
Statistical analysis
Data are presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) and two-tailed Student’s t-test was used to analyze statistical differences between the mean values. P < 0.05 was considered as statistically significant.
Results
Chromatography and mass spectrometry analysis for neratinib in rat plasma
The UPLC-MS/MS conditions were initially optimized for maximum sensitivity and resolution. It was found that addition of 2.5 mM ammonium acetate in mobile phase A (water) could significantly improve peak shape and increase signal strength. The addition of 0.1% of formic acid to both mobile phases was found to be essential for promoting the ionization of both analytes, which further increased detection sensitivity. Neratinib and IS (sorafenib) were detected both under positive and negative ion mode, and the positive mode was finally selected because the analytes were more efficiently ionized. In the positive ion mode, neratinib and IS showed a protonated molecular ion at m/z 557.50 and 465.31 (Fig. 1), respectively. Parameter optimization of mass spectrometer settings revealed the most abundant daughter ions were m/z 111.98 for neratinib (Fig. 1A) at a collision energy of 30 V and 252.11 for IS (Fig. 1B) at a collision energy of 35 V. Hence, ion transitions for quantification were m/z 557.50 > 111.98 for neratinib and m/z 465.31 > 252.11 for IS monitored using the MRM technique (Fig. 1).

Parent and Daughter ion transition spectra of neratinib A and IS B in rat plasma
Method validation
Specificity
The MRM chromatograms of plasma with the analyte and IS show no interference at the respective retention regions of neratinib (1.96 min) and IS (2.91 min) (Fig. 2A and B). The result indicated that the method for neratinib quantification was highly selective.

Representative MRM chromatograms of neratinib and IS in a prepared rat plasma sample. The results showed the molecular ion m/z 557/112 for neratinib A and m/z 465/252 for IS B. C A plasma sample spiked with a standard mixture of neratinib at its LLOQ level
Linearity and LLOQ
Neratinib showed good linearity in the concentration range of 20-4000 ng/mL (y = 0.0115x + 0.336) with the correlation coefficient (R2) of > 0.999. The LLOQ of neratinib in plasma was 2 ng/mL (Fig. 2C).
Accuracy and precision
The assay for neratinib was validated with an acceptable intra-day and inter-day accuracies (n = 6) ranged between 99.14% and 102.96% (Table1). The CV% values were with 10% during the intra-day and inter-day precision study for all concentrations (Table 1) and conformed to the accepted variable limits.
Extraction recoveries and matrix effects
The extraction recoveries of neratinib in plasma for three concentrations (40 ng/mL, 400 ng/mL and 4000 ng/mL) were not less than 90% (Table 2). The matrix effects ranged from 103.80% to 107.35% for neratinib in plasma for all concentrations (Table 2). The CV% for extraction recoveries and matrix effects were within 10% (Table 2), respectively. These results suggested an optimum extraction assay and negligible matrix effect for analysis of neratinib in plasma.
Stability
The stabilities of neratinib (40 ng/mL, 400 ng/mL and 4000 ng/mL) under different stressing conditions were evaluated. As shown in Table 3, %recoveries of neratinib for all concentrations in plasma at -80 °C for 10 days and after three freeze–thaw cycles were between 98.78% and 103.03%. In addition, we also determined the stabilities of neratinib in 50% acetonitrile solution at 4 °C for 24 h and ambient temperature for 4 h. The %recoveries of all samples were not less than 96%. All of the above results indicated neratinib was stable under the studied stress conditions.
Effects of verapamil on the pharmacokinetics of neratinib in rats
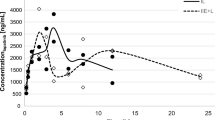
The validated UPLC-MS/MS method was used to determine the oral pharmacokinetics of neratinib in Sprague–Dawley rats with or without pretreatment of verapamil. The mean plasma concentration–time curves of neratinib in the two groups were depicted in Fig. 3 and the pharmacokinetic parameters were presented in Table 4. After pretreatment with verapamil, the area under the plasma concentration–time curve (AUC0-t) of neratinib significantly increased by 1.64-fold (P < 0.05). The maximum plasma concentration (Cmax) of neratinib showed a significant increase by 2.09-fold (P < 0.001) and the time to reach it (Tmax) was earlier in pretreatment group rats compared to control group rats. However, the half time (t1/2) was not significantly different between the two groups. Further, the Vd/F and CL/F decreased significantly from 26.86 to 14.63 L/kg (P < 0.05) and 2.70 to 1.66 L/kg/h (P < 0.05), respectively. These results indicated the systemic exposure of neratinib in rats was increased when administered in combination with verapamil.

Plasma concentration–time profile of neratinib (100 mg/kg) in rats after an oral administration with or without verapamil (10 mg/kg) pretreatment. Each point is shown as the average concentration with the error bar representing the S.D. (n = 5)
Intracellular accumulation of neratinib in MDCK II and MDCK-MDR1 cells
Western blotting results showed that P-gp was successfully overexpressed in MDCK-MDR1 cells compared to MDKC II cells (Fig. 4A). At both high (5 μM) and low (1.25 μM) dosing concentrations, the intracellular amount of neratinib in MDCK II cells was significant higher (P < 0.001) than that in MDCK-MDR1 cells (Fig. 4B). Further, the effect of verapamil (100 μM) on the intracellular accumulation was determined. As shown in Fig. 4C, verapamil did not alter the accumulation of neratinib in MDCKII cells. By contrast, the intracellular amount of neratinib in MDCK-MDR1 cells was markedly increased (P < 0.001) by verapamil (Fig. 4D). Taken together, this result indicated that P-gp might be involved in the transport of neratinib and verapamil could inhibit the efflux of neratinib in MDCK-MDR1 cells.

(A) Protein expression of P-gp in MDCK II and MDCK-MDR1 cells determined by Western blotting. (B) Intracellular accumulation of neratinib (1.25 and 5 μΜ) in MDCK II and MDCK-MDR1 cells. (C) Effect of verapamil (100 µM) on the intracellular accumulation of neratinib (1.25 and 5 μΜ) in MDCK II cells. (D) Effect of verapamil (100 µM) on the intracellular accumulation of neratinib (1.25 and 5 μΜ) in MDCK-MDR1 cells. (E) Effect of verapamil (100 µM) on the transport of neratinib (5 μΜ) from the apical to basolateral side or the opposite direction in MDCK-MDR1 monolayers. Each bar is the average of three determinations with the error bar representing the S.D. (n = 3). ***P < 0.001 versus the control group. ###P < 0.001 compared Papp, BL-AP with Papp,AP-BL
Transport of neratinib across MDCK II and MDCK-MDR1 cell monolayers
To further investigate whether neratinib was a substrate of P-gp, the MDCK II and MDCK-MDR1 cell monolayers were utilized. As shown in Fig. 4E and Table 5, neratinib exhibited directional transport in MDCK-MDR1 cells. The Papp value of neratinib from basal-to-apical (Papp, BL-AP) was 24.24 ± 2.75 × 10–6 cm/s, which was significant higher (P < 0.001) than that (9.84 ± 0.66 × 10–6 cm/s) form apical-to-basal (Papp, AP-BL). The efflux ration was further calculated as 2.46 (> 2) based on Papp, BL-AP divided by Papp, AP-BL. When verapamil was used, the Papp, BL-AP of neratinib was significantly decreased to 15.60 ± 0.04 × 10–6 cm/s, as the efflux ratio decreased to 1.01. As shown in Table 5, preincubation with verapamil resulted in a decrease in the net flux ratio of 79.6% for neratinib. These results indicated that neratinib was identified as a P-gp substrate with a net flux ratio reduced more than 50% by verapamil, attesting to the specificity of P-gp mediated efflux.
Discussion
In this study, we have constructed a highly specific, selective and sensitive UPLC-MS/MS method for the simultaneous quantification of neratinib and IS (sorafenib) in rat plasma (Figs. 1 and 2, Tables 1, 2 and 3). This validated UPLC-MS/MS assay was successfully applied to the pharmacokinetic determination of neratinib in rats. Our findings have demonstrated that pre-treatment with verapamil (a known P-gp inhibitor) resulted in significant increase in system exposure of neratinib in rats, as the value of Cmax and AUC0-t increased and CL/F of neratinib decreased (Fig. 3 and Table 4). Further, our study was the first one to confirmed that P-gp was involved in the transport of neratinib using MDCK II and MDCK-MDR1 cells model (Fig. 4 and Table 5).
Neratinib is currently approved for the treatment of patients with early-stage and metastatic HER2+ breast cancer [1]. However, it is estimated that about 70% of HER2+ breast cancer patients are either innately resistant or acquire resistance to HER2-targeted drugs [18, 19]. Abbas et al. have identified neratinib as a sensitive substrate for CYP3A4 enzyme, and co-administration with CYP3A4 inhibitor caused significant increased system exposure of neratinib [18, 20, 21]. Therefore, increased CYP3A4 activity has been considered to be an important mechanism of neratinib resistance. In addition to the action of metabolic enzymes, drug excretion mediated by efflux transporters, overexpressed in tumor cells, also confers to the commonly known phenomenon of multidrug resistance against certain antineoplastic agents [22, 23]. P-gp has been known to transport a wide range of drugs and xenobiotics from intra- to extracellular at many biological interfaces such as the intestine, liver, blood-brain barrier (BBB), and kidney. Hence, activity inhibition of P-gp may raise blood concentrations of its substrates. In fact, we have evidenced that pre-treatment with verapamil significantly increased neratinib Cmax compared with administration of neratinib alone (Table 4), resulting in higher plasma exposure level of neratinib (Fig. 3). Verapamil has been reported as an inhibitor of both CYP3A4 and P-gp and used for drug-drug interaction studies in vitro and in vivo [24, 25]. Therefore, we inferred that verapamil may affect neratinib exposure partially via inhibiting the activity of CYP3A4. However, whether P-gp inhibition by verapamil can affect the in vivo pharmacokinetics of neratinib should be further investigated.
Thus, MDCK cells transfected with the human MDR1 gene (MDCK-MDR1) cells were used to examine the influence of P-gp on the active transport of neratinib. In the intracellular accumulation assay, verapamil successfully inhibited the efflux of neratinib in MDCK-MDR1 cells (Fig. 4D). In transcellular bidirectional transport experiments, neratinib has been identified as a P-gp substrate according to the net flux ratio (> 2) (Fig. 4E and Table 5). Hence, induction or inhibition of P-glycoprotein can lead to drug-drug interactions of neratinib. The changed pharmacokinetics of neratinib in rats may partially due to P-gp inhibition by verapamil. Further, due to the overexpression of P-gp in tumor cells, the multidrug resistance (MDR) function of P-gp would affect the clinical therapeutic effect of neratinib, which may be a crucial mechanism for neratinib resistance.
Additionally, breakthroughs in cancer research have greatly improved the survival of patients with advanced breast cancer, however, the incidence of brain metastases has also increased accordingly [12, 26]. Significantly, the HER2+ subtype harbored higher risk to metastasize to the brain than other breast cancer subtypes [26]. Neratinib, to a degree, has shown intracranial efficacy in treatment for HER2+ breast cancer patients with brain metastases. But many challenges remain as the barrier properties of BBB, resulting in the sub-therapeutic concentrations of chemotherapeutic drugs within the brain. P-gp is expressed at high levels in the capillary endothelial cells of the BBB, and activity inhibition of P-gp can result in increased concentration of substrate drugs [27,28,29]. Taken together, the therapeutic effect of neratinib in treating brain metastases will likely to be improved when co-administrated with P-gp inhibitors. However, further research is needed to verify this suppose.
Furthermore, the mechanism for neratinib efflux in BBB is not only related to P-glycoprotein, but also may involve breast cancer resistance protein (BCRP), which is also located at the apical membranes of BBB [29, 30]. These two transporters act cooperatively at the BBB, limiting the uptake of substrate drugs into the central nervous system (CNS) and affecting their pharmacokinetics, therapeutic efficacy, and safety [31, 32]. In fact, we have investigated the role of BCRP on the transport of neratinib using MDCK II and stably transfected lines with human ABCG2 genes (MDCK-BCRP). However, the intracellular accumulation of neratinib in MDCK-BCRP cells showed no statistically significant difference with that in MDCK II cells. The addition of BCRP inhibitor (10 μM Ko143) did not alter the efflux of neratinib in MDCK-BCRP cells, suggesting that neratinib was probably not substrate for BCRP. Thus, the absorption of neratinib and its tissue distribution (i.e., brain) is considered mainly mediated by the activity or expression of P-gp protein.
MDCK cells belong to epithelial cell line of canine kidney origin, and are increasing used in evaluation of drug passive permeability and membrane transport studies, and easy to handle [33]. MDCK-MDR1 cells provides a suitable in vitro model for screening P-gp substrates, showing polarized overexpression of P-gp in the apical side [33, 34]. The results obtained in MDCK cell well reflect the in vivo situation [31, 34]. In addition, bidirectional transport assays with MDCK-MDR1 cells have been reported as a valuable in vitro assay to investigate if human P-gp prevents the test compound from crossing the BBB [35,36,37]. Therefore, the results obtained by MDCK II and MDCK-MDR1 cell lines can reveal the influence of P-gp on the access of neratinib to the CNS. However, the pharmacokinetics of neratinib in brain need to be further investigated in future studies.
Conclusion
In conclusion, our results indicated that the system exposure of neratinib would be significantly increased when the rats were pre-treatment with verapamil. Furthermore, neratinib was identified as a substrate of P-gp using the in vitro assays with MDCK II and MDCK-MDR1 cells, revealing the main mechanism underling the effect of verapamil on neratinib absorption in rats. To the best of our knowledge, thus far, there are no published in vitro or in vivo studies characterizing the role of P-gp on the transport of neratinib. What’s more, in light of potential application of neratinib in HER2+ breast cancer with brain metastases, our findings may provide relevant information for the factors that govern the access of neratinib to the CNS, especially for the treatment of (metastatic) brain cancers. However, additional investigation should be performed to validate these results.
Abbreviations
- BBB:
-
Blood-brain barrier
- BCRP:
-
Breast cancer resistance protein
- ER:
-
Efflux ratio
- HER:
-
Human epidermal receptor
- IS:
-
Internal standard
- LOQ:
-
Limit of quantification
- MDCK:
-
Madin-Darby canine kidney
- Papp :
-
Apparent permeability
- P-gp:
-
P-glycoprotein
- TEER:
-
Transepithelial electrical resistance
- UPLC-MS/MS:
-
Ultra-performance liquid chromatography-tandem mass spectrometry
References
Deeks ED (2017) Neratinib: First Global Approval. Drugs 77(15):1695–1704. https://doi.org/10.1007/s40265-017-0811-4
Barcenas CH, Hurvitz SA, Di Palma JA, Bose R, Chien AJ, Iannotti N, Marx G, Brufsky A, Litvak A, Ibrahim E, Alvarez RH, Ruiz-Borrego M, Chan N, Manalo Y, Kellum A, Trudeau M, Thirlwell M, Garcia Saenz J, Hunt D, Bryce R, McCulloch L, Rugo HS, Tripathy D, Chan A, Investigators CS (2020) Improved tolerability of neratinib in patients with HER2-positive early-stage breast cancer: the CONTROL trial. Ann Oncol 31(9):1223–1230. https://doi.org/10.1016/j.annonc.2020.05.012
Tiwari SR, Mishra P, Abraham J (2016) Neratinib, A Novel HER2-Targeted Tyrosine Kinase Inhibitor. Clin Breast Cancer 16(5):344–348. https://doi.org/10.1016/j.clbc.2016.05.016
Chila G, Guarini V, Galizia D, Geuna E, Montemurro F (2021) The Clinical Efficacy and Safety of Neratinib in Combination with Capecitabine for the Treatment of Adult Patients with Advanced or Metastatic HER2-Positive Breast Cancer. Drug Des Devel Ther 15:2711–2720. https://doi.org/10.2147/DDDT.S281599
Cunningham N, Shepherd S, Mohammed K, Lee KA, Allen M, Johnston S, Kipps E, McGrath S, Noble J, Parton M, Ring A, Turner NC, Okines AFC (2022) Neratinib in advanced HER2-positive breast cancer: experience from the royal Marsden hospital. Breast Cancer Res Treat. https://doi.org/10.1007/s10549-022-06703-3
Martin M, Holmes FA, Ejlertsen B, Delaloge S, Moy B, Iwata H, von Minckwitz G, Chia SKL, Mansi J, Barrios CH, Gnant M, Tomasevic Z, Denduluri N, Separovic R, Gokmen E, Bashford A, Ruiz Borrego M, Kim SB, Jakobsen EH, Ciceniene A, Inoue K, Overkamp F, Heijns JB, Armstrong AC, Link JS, Joy AA, Bryce R, Wong A, Moran S, Yao B, Xu F, Auerbach A, Buyse M, Chan A, Exte NETSG (2017) Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 18(12):1688–1700. https://doi.org/10.1016/S1470-2045(17)30717-9
Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, Reich MF, Shen R, Shi X, Tsou HR, Wang YF, Wissner A (2004) Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res 64(11):3958–3965. https://doi.org/10.1158/0008-5472.CAN-03-2868
Zhao M, Scott S, Evans KW, Yuca E, Saridogan T, Zheng XF, Wang HP, Korkut A, Pico CXC, Demirhan M, Kirby B, Kopetz S, Diala I, Lalani AS, Piha-Paul S, Meric-Bernstam F (2021) Combining Neratinib with CDK4/6, mTOR, and MEK Inhibitors in Models of HER2-positive Cancer. Clin Cancer Res 27(6):1681–1694. https://doi.org/10.1158/1078-0432.Ccr-20-3017
Li Y, Yuan H, Yang K, Xu W, Tang W, Li X (2010) The structure and functions of P-glycoprotein. Curr Med Chem 17(8):786–800. https://doi.org/10.2174/092986710790514507
Lin JH, Yamazaki M (2003) Clinical relevance of P-glycoprotein in drug therapy. Drug Metab Rev 35(4):417–454. https://doi.org/10.1081/dmr-120026871
Waghray D, Zhang Q (2018) Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J Med Chem 61(12):5108–5121. https://doi.org/10.1021/acs.jmedchem.7b01457
Corti C, Antonarelli G, Criscitiello C, Lin NU, Carey LA, Cortes J, Poortmans P, Curigliano G (2022) Targeting brain metastases in breast cancer. Cancer Treat Rev 103:102324. https://doi.org/10.1016/j.ctrv.2021.102324
Stemmler HJ, Schmitt M, Willems A, Bernhard H, Harbeck N, Heinemann V (2007) Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2-positive breast cancer patients with brain metastases and impairment of blood-brain barrier. Anti-Cancer Drug 18(1):23–28. https://doi.org/10.1097/01.cad.0000236313.50833.ee
Shah N, Mohammad AS, Saralkar P, Sprowls SA, Vickers SD, John D, Tallman RM, Lucke-Wold BP, Jarrell KE, Pinti M, Nolan RL, Lockman PR (2018) Investigational chemotherapy and novel pharmacokinetic mechanisms for the treatment of breast cancer brain metastases. Pharmacol Res 132:47–68. https://doi.org/10.1016/j.phrs.2018.03.021
Ni J, Wang YZ, Diala I, Kabraji S, Freedman R, Lin N, Zhao J (2019) Preclinical evaluation of neratinib plus T-DM1 in orthotopic PDX models of HER2-positive breast cancer brain metastases. Cancer Res 79(13). https://doi.org/10.1158/1538-7445.Am2019-4832
Freedman RA, Gelman RS, Agar NYR, Santagata S, Randall EC, Gimenez-Cassina Lopez B, Connolly RM, Dunn IF, Van Poznak CH, Anders CK, Melisko ME, Silvestri K, Cotter CM, Componeschi KP, Marte JM, Moy B, Blackwell KL, Puhalla SL, Ibrahim N, Moynihan TJ, Nangia J, Tung N, Burns R, Rimawi MF, Krop IE, Wolff AC, Winer EP, Lin NU, Translational Breast Cancer Research C (2020) Pre- and Postoperative Neratinib for HER2-Positive Breast Cancer Brain Metastases: Translational Breast Cancer Research Consortium 022. Clin Breast Cancer 20(2):145–151 e142. https://doi.org/10.1016/j.clbc.2019.07.011
Zhang XJ, Liu T, Zhang YD, Liu FY, Li HY, Fang D, Wang CJ, Sun H, Xie SQ (2019) Elucidation of the Differences in Cinobufotalin’s Pharmacokinetics Between Normal and Diethylnitrosamine-Injured Rats: The Role of P-Glycoprotein. Front Pharmacol. https://doi.org/10.3389/fphar.2019.00521
Breslin S, Lowry MC, O’Driscoll L (2017) Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Brit J Cancer 116(5):620–625. https://doi.org/10.1038/bjc.2016.445
Velaga R, Tanaka S, Toi M (2022) Molecular vulnerabilities and therapeutic resistance in hormone receptor positive and HER2 dependent breast cancer tumours. Cancer Drug Resist 5(2):487–497. https://doi.org/10.20517/cdr.2022.10
Abbas R, Hug BA, Leister C, Burns J, Sonnichsen D (2011) Pharmacokinetics of oral neratinib during co-administration of ketoconazole in healthy subjects. Br J Clin Pharmacol 71(4):522–527. https://doi.org/10.1111/j.1365-2125.2010.03845.x
Yu J, Petrie ID, Levy RH, Ragueneau-Majlessi I (2019) Mechanisms and Clinical Significance of Pharmacokinetic-Based Drug-Drug Interactions with Drugs Approved by the U.S. Food and Drug Administration in 2017. Drug Metab Dispos 47(2):135–144. https://doi.org/10.1124/dmd.118.084905
Rachmale M, Rajput N, Jadav T, Sahu AK, Tekade RK, Sengupta P (2022) Implication of metabolomics and transporter modulation based strategies to minimize multidrug resistance and enhance site-specific bioavailability: a needful consideration toward modern anticancer drug discovery. Drug Metab Rev 54(2):101–119. https://doi.org/10.1080/03602532.2022.2048007
Theodoulou FL, Kerr ID (2015) ABC transporter research: going strong 40 years on. Biochem Soc T 43:1033–1040. https://doi.org/10.1042/Bst20150139
Elbadawy HA, Wahdan SA, El-Demerdash E (2022) Effect of Atorvastatin on Single Oral Pharmacokinetics and Safety of Daclatasvir in Rats: Emphasis on P-glycoprotein and Cytochrome P450. Curr Drug Metab 23(6):484–495. https://doi.org/10.2174/1389200223666220404134524
Srinivas NR (2008) Dual drug interactions via P-glycoprotein (P-gp)/ cytochrome P450 (CYP3A4) interplay: recent case study of oral atorvastatin and verapamil. Eur J Clin Pharmacol 64(11):1135–1136. https://doi.org/10.1007/s00228-008-0512-8
Meattini I, Andratschke N, Kirby AM, Sviri G, Offersen BV, Poortmans P, Kaidar Person O (2020) Challenges in the treatment of breast cancer brain metastases: evidence, unresolved questions, and a practical algorithm. Clin Transl Oncol 22(10):1698–1709. https://doi.org/10.1007/s12094-020-02333-7
Karbownik A, Sobanska K, Plotek W, Grabowski T, Klupczynska A, Plewa S, Grzeskowiak E, Szalek E (2020) The influence of the coadministration of the p-glycoprotein modulator elacridar on the pharmacokinetics of lapatinib and its distribution in the brain and cerebrospinal fluid. Invest New Drugs 38(3):574–583. https://doi.org/10.1007/s10637-019-00806-3
Fm ML, Loos NHC, Mucuk S, de Jong D, Lebre MC, Rosing H, Tibben M, Beijnen JH, Schinkel AH (2021) P-Glycoprotein (ABCB1/MDR1) Controls Brain Penetration and Intestinal Disposition of the PARP1/2 Inhibitor Niraparib. Mol Pharm 18(12):4371–4384. https://doi.org/10.1021/acs.molpharmaceut.1c00553
de Vries NA, Buckle T, Zhao J, Beijnen JH, Schellens JHM, van Tellingen O (2012) Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and BCRP. Invest New Drug 30(2):443–449. https://doi.org/10.1007/s10637-010-9569-1
Goncalves J, Silva S, Gouveia F, Bicker J, Falcao A, Alves G, Fortuna A (2021) A combo-strategy to improve brain delivery of antiepileptic drugs: Focus on BCRP and intranasal administration. Int J Pharm 593:120161. https://doi.org/10.1016/j.ijpharm.2020.120161
Bicker J, Fortuna A, Alves G, Soares-da-Silva P, Falcao A (2017) Elucidation of the Impact of P-glycoprotein and Breast Cancer Resistance Protein on the Brain Distribution of Catechol-O-Methyltransferase Inhibitors. Drug Metab Dispos 45(12):1282–1291. https://doi.org/10.1124/dmd.117.077883
Hernandez-Lozano I, Mairinger S, Traxl A, Sauberer M, Filip T, Stanek J, Kuntner C, Wanek T, Langer O (2021) Assessing the Functional Redundancy between P-gp and BCRP in Controlling the Brain Distribution and Biliary Excretion of Dual Substrates with PET Imaging in Mice. Pharmaceutics 13(8). https://doi.org/10.3390/pharmaceutics13081286
Volpe DA (2011) Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med Chem 3(16):2063–2077. https://doi.org/10.4155/fmc.11.149
Hu HH, Bian YC, Liu Y, Sheng R, Jiang HD, Yu LS, Hu YZ, Zeng S (2014) Evaluation of blood-brain barrier and blood-cerebrospinal fluid barrier permeability of 2-phenoxy-indan-1-one derivatives using in vitro cell models. Int J Pharmaceut 460(1–2):101–107. https://doi.org/10.1016/j.ijpharm.2013.11.013
Brouwer KL, Keppler D, Hoffmaster KA, Bow DA, Cheng Y, Lai Y, Palm JE, Stieger B, Evers R, International Transporter C (2013) In vitro methods to support transporter evaluation in drug discovery and development. Clin Pharmacol Ther 94(1):95–112. https://doi.org/10.1038/clpt.2013.81
Yang B, Du S, Lu Y, Jia S, Zhao M, Bai J, Li P, Wu H (2018) Influence of paeoniflorin and menthol on puerarin transport across MDCK and MDCK-MDR1 cells as blood-brain barrier in vitro model. J Pharm Pharmacol 70(3):349–360. https://doi.org/10.1111/jphp.12853
Palmer AM, Alavijeh MS (2013) Overview of experimental models of the blood-brain barrier in CNS drug discovery. Curr Protoc Pharmacol 62:715 11–17 15 30. https://doi.org/10.1002/0471141755.ph0715s62
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81703801), National Key R&D Program of China (2021YFA0909600), Key Project of Science and Technology Research Funded by the Educational Commission of Henan Province (22A350013), the Key Technologies R&D Program of Henan (212102311108, 222102310397, 202102310405, 222102310216), and Key Program “New Drug Creation” of Guangdong Key Research and Development Plan (2019B020202001).
Author information
Authors and Affiliations
Contributions
Participated in research design: Defei Qi, Yuanyuan Dou and Hua Sun. Conducted experiments: Defei Qi, Yuanyuan Dou, Wenke Zhang, Mengqing Wang, Yingying Li, Mingzhu Zhang, Jia Qin, Jinlan Cao and Hua Sun. Contributed new reagents or analytic tools: Dong Fang, Jing Ma, Wei Yang and Songqiang Xie. Performed data analysis: Defei Qi, Yuanyuan Dou and Hua Sun. Wrote or contributed to the writing of the manuscript: Defei Qi and Hua Sun.
Corresponding authors
Ethics declarations
Conflict of interests
The authors have declared no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qi, D., Dou, Y., Zhang, W. et al. The influence of verapamil on the pharmacokinetics of the pan-HER tyrosine kinase inhibitor neratinib in rats: the role of P-glycoprotein-mediated efflux. Invest New Drugs 41, 13–24 (2023). https://doi.org/10.1007/s10637-022-01314-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-022-01314-7