
Summary
Introduction This Phase Ib trial investigated the safety, tolerability, and recommended phase 2 dose for the pan-PI3K/mTOR inhibitor, GSK2126458 (GSK458), and trametinib combination when administered to patients with advanced solid tumors. Patients and Methods Patients with advanced solid tumors received escalating doses of GSK458 (once or twice daily, and continuous or intermittent) and trametinib following a zone-based 3 + 3 design to determine the maximum tolerated dose (MTD). Assessments included monitoring for adverse events and response, and evaluating pharmacokinetic (PK) measures. Archival tissue and circulating free DNA samples were collected to assess biomarkers of response in the PI3K and RAS pathways. Results 57 patients were enrolled onto the continuous dosing cohort and 12 patients onto an intermittent BID dosing cohort. Two MTDs were established for the continuous daily dosing: 2 mg of GSK458 with 1.0 mg of trametinib or 1.0 mg of GSK458 with 1.5 mg of trametinib; no MTD was determined in the intermittent dosing cohort. The most frequent adverse events were rash (74 %) and diarrhea (61 %). Dose interruptions due to adverse events occurred in 42 % of patients. No significant PK interaction was observed. One patient achieved partial response and 12 patients had stable disease >16 weeks. Mutations in RAS/RAF/PI3K were detected in 70 % of patients, but no pattern emerged between response and mutational status. Conclusion GSK458 plus trametinib is poorly tolerated, due to skin and GI-related toxicities. Responses were minimal, despite enrichment for PI3K/RAS pathway driven tumors, which may be due to overlapping toxicities precluding sufficient dose exposure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Components of the phosphoinositide 3-kinase (PI3K)/AKT and RAS/RAF/MEK pathways are frequently co-mutated or aberrantly co-expressed in human cancers leading to proliferation, survival and treatment resistance [1]. Synergistic anti-tumor effects have been observed in preclinical models when inhibiting both pathways simultaneously [2–5].
GSK2126458 (GSK458) is a potent, reversible dual pan-PI3K and mammalian target of rapamycin (mTOR) inhibitor. The maximum tolerated dose (MTD) of GSK458 was established at 2.5 mg once daily. Twice daily (BID) dosing was later selected as the preferred administration schedule because of a more favorable pharmacokinetic (PK) profile. The most frequently reported treatment-related adverse events (AEs) observed were diarrhea, fatigue, nausea, decreased appetite, vomiting, and hyerpglycemia [6]. Trametinib (Mekinst®) is a reversible, selective inhibitor of MEK1/MEK2 kinase that is approved for the treatment of metastatic or unresectable BRAF V600E/K mutant melanoma. The recommended phase II dose (RP2D) of trametinib is 2 mg once daily. The most frequently reported treatment-related toxicities are rash or acneiform dermatitis, diarrhea, fatigue, nausea, and vomiting [7]. In vitro synergy for cell growth inhibition is observed with the GSK458 and trametinib combination in breast, colorectal, lung, and pancreatic cancer cell lines (GSK data on file).
The primary objective of this Phase Ib trial was to determine the safety, tolerability, and RP2D for the GSK458 and trametinib combination when administered to patients with advanced solid tumors. Secondary objectives were to evaluate PK, pharmacodynamic effects, and clinical activity of the combination.
Patients and methods
Study population
Patient age ≥ 18 years with relapsed or refractory solid tumors who provided written consent, had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and adequate hematologic and organ function were enrolled. Key exclusion criteria were: treatment with anti-cancer therapy within 28 days, history of retinal vein occlusion or central serous retinopathy or visible retinal pathology, diabetes (type 1 or 2), uncontrolled systemic diseases, unstable brain metastases, history of major cardiovascular disease, and/or pregnant/lactating females. To enrich for patients with increased likelihood to derive clinical benefit, the study was amended to specifically enroll KRAS-mutant colorectal cancer (CRC), KRAS-mutant non-small cell lung cancer (NSCLC), BRAF-mutant melanoma previously treated with a BRAF inhibitor, pancreatic, endometrial, ovarian, bladder, triple negative breast cancer, and other cancers with a rationale for co-treatment with PI3K and MEK inhibitors. Prior therapy with PI3K and MEK pathway inhibitors was permitted.
All relevant Institutional Review Boards approved the protocol, in compliance with the recommendations of the Helsinki Declaration. Written informed consent was obtained from each patient before enrollment.
Study design
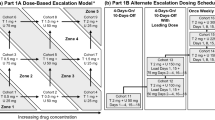
This open-label, multicenter study (NCT01248858) evaluated three administration schedules: continuous GSK458 once daily plus trametinib once daily for 28 days (cohorts 1–9), continuous GSK458 twice daily plus trametinib once daily for 28 days (cohort 10), and intermittent GSK458 BID (4 days on, 10 days off) plus trametinib once daily for 28 days (cohorts 11 and 12). Patients fasted for 1 h before and 2 h after dosing of both study medications. Dose escalation followed a zone-based design (3 + 3) with rules for continuous and intermittent dosing of GSK458 together with continuous daily trametinib dosing (Fig. 1). A conservative starting dose of 0.5 mg once daily for both drugs (~70 % reduction) was based upon potential overlapping toxicities of diarrhea, rash, and possible cardiac toxicities.

Summary of dose escalation design and tolerability. a continuous daily dosing schedules, b intermittent dosing schedules. BID=twice daily; GSK468=GSK212458; MTD=maximum tolerated dose; QD=daily; T=trametinib
Safety and efficacy assessments
Patients were assessed for safety on days 1, 8, 15, 29, 43, and then every 4 weeks. Clinical monitoring included routine chemistry, hematology, vital signs, echocardiogram (monthly), electrocardiogram, physical examination, ophthalmic examination (baseline then as needed), and AE assessment per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) criteria (version 4.0). Additional safety evaluations included daily home fingerstick blood glucose, and intermittent monitoring of fasting blood glucose and insulin levels, hemoglobin A1C, 1,5-anhydroglucitol, C-peptide, fasting lipid panel, and troponin. Dose limiting toxicities (DLT) definitions included grade 4 hematologic toxicities or febrile neutropenia, and most grade 3 non-hematologic toxicities that occurred in the first 28-day treatment period. Exceptions included grade 3 or greater nausea, vomiting, diarrhea, mucositis/esophagitis that responded to standard supportive treatment(s) within 48 h, electrolyte disturbances that responded to correction within 24 h, and asymptomatic grade 3 hypertension that was adequately controlled by the addition of up to two additional antihypertensive medications. Additional DLT criteria included grade 3 rash that did not improve to grade ≤ 2 within 7 days despite optimal supportive treatment(s) or required a dose reduction upon restarting therapy, ALT >3× upper limit of normal (ULN) with concurrent bilirubin >2× ULN, absolute decrease of ejection fraction to less than lower limit of normal and >10 % decrease from baseline confirmed within 7 days, inability to receive ≥75 % of scheduled doses due to toxicity, or grade 2 or higher toxicity that occurred beyond 28 days at the judgment of the investigator and medical monitor. The MTD was defined as the highest dose at which ≤1/6 patients experienced a DLT in the first 28 days of therapy. Anti-tumor efficacy was assessed by CT/MRI at week 6 and then every 8 weeks based on clinical evaluation and RECIST 1.1 criteria [8].
Pharmacokinetic considerations
Two sparse PK samples were collected from all patients in the study pre-dose and 1–3 h post-dose on Days 29, 43, 99 of continuous dosing. For the GSK458 alternate dose schedule of 4 day on/10 day off, PK samples were collected pre-dose and at 1, 2, 4 h on days 1, 4, 15, and predose, 1, 2, 4, 6, 8, 24 h on day 18, and predose days 29, 43, 99.
Population PK (POP PK) analysis using NONMEM software (version 7.2) was performed both analytes (GSK458 and trametinib) to characterize the PK of GSK458 alone and in combination with trametinib to assess for possible interaction. A two-compartment model with first order absorption and elimination was used to describe concentration time profile of GSK458. First-order conditional estimation method with interaction was applied for the model development as well as covariate identification. The mean population clearance, inter compartment clearance, volume of distribution in central and peripheral compartment, absorption rate constant, inter-subject variability as well as residual error were estimated. Final model selection was based on evaluation of goodness-of-fit plots, biological plausibility, precision of parameter estimates, and the minimum objective function value. Covariates affecting PK of GSK458 were also explored.
Archival tumor and circulating free DNA (cfDNA) characterization
Archival formalin-fixed paraffin embedded tumor samples were collected to assess biomarkers of response (eg, loss of PTEN, PIK3CA, and RAS mutations). Pre-treatment plasma samples were characterized for somatic mutations using circulating free DNA (cfDNA) [9] in the continuous daily dosing cohort to explore the relationship between cfDNA and anti-tumor activity.
Statistical analysis
Dose escalation decisions were based on synthesis of all relevant available clinical, PK, and laboratory data and not solely on DLT information. Data were modeled using an adaptive 4-parameter Bayesian logistic regression model (BLRM) with overdose control principle [10]. The BLRM was fitted on the DLT data accumulated throughout the dose-escalation in the first 28 days to model the dose-toxicity relationship of GSK458 and trametinib when given in combination. All available dose-DLT relationships of single agents GSK458 and trametinib were included in the BLRM fitting with 10 % weight. A non-informative Jeffrey’s prior was used for the model parameters. [11, 12].
Results
Patient characteristics and treatment delivered
From 03 December 2010 to 28 August 2013, 57 patients were enrolled into the continuous dosing cohort and 12 patients to an intermittent BID dosing cohort (Table 1). The database was locked on 01 May 2014. The study included a range of tumor types with more than half being KRAS-mutant colorectal, breast, KRAS-mutant NSCLC, or pancreatic cancer. The median time on treatment was 52 days (range: 14–492 days) for the continuous dosing cohort and 72 days (range: 35–357 days) for the intermittent dosing cohort.
Dose escalation and DLTs
Eight DLTs were observed across the 12 dose levels explored (Table 2). At 1 mg of trametinib with 1 mg daily GSK458, 1/6 patients had a DLT of G2 folliculitis requiring a 20 day interruption in study treatment; with 1.5 mg GSK458 daily 1/8 patients had a DLT of G3 diarrhea; at 2 mg GSK458 0/7 patients had DLTs. 1.5 mg of trametinib could be given safely with 1 mg GSK458, but 2 mg of trametinib with 0.5 mg of GSK458 exceeded the MTD with 3/5 patients experiencing DLTs of G3 stomatitis, G3 hypertension, and G3 hypophosphatemia with G2 diarrhea. Therefore two MTDs were established for continuous daily dosing: 2 mg of GSK458 with 1.0 mg of trametinib or 1.0 mg of GSK458 with 1.5 mg of trametinib.
BID dosing of GSK458 was also explored to allow sustained exposures across the majority of the 24 h dosing period. At the first dose level, 1.5 mg of trametinib with 0.5 mg BID GSK458 exceeded the MTD with two DLTs of G3 mucositis and an asymptomatic 20 % decrease in LVEF (confirmed with 7 day follow up). Due to inability to achieve full monotherapy doses for both agents on continuous dosing schedules, two cohorts of intermittent dosing (int GSK458 at 0.5 mg BID and 0.75 mg, administered twice daily for four days on and ten days off) were explored in combination with continuous dosing of trametinib at 1.5 mg QD. At the 0.5 mg BID int GSK458 dose, 1/7 patients experienced a G3 DLT of central serous retinopathy. Although all five patients dosed cleared the 0.75 mg BID int GSK458 dose, the study was terminated since neither continuous nor intermittent exposures to GSK458 that had been predicted as being necessary for on target activity of the combination were anticipated to be achievable.
Safety and tolerability
AEs across all dose combinations irrespective of causality are shown in Table 3 There were no treatment related grade 4 or 5 AEs. Rash (74 %) and diarrhea (61 %) were the most frequent all grade toxicities, and the main treatment-related grade 3 AEs occurring in 7 (10 %) and 4 (6 %) of the 69 patients respectively. Other common all-cause AEs included vomiting (45 %), fatigue (39 %), nausea (35 %), mucositis (35 %), and peripheral edema (33 %). Although only 12 patients were treated in the intermittent dosing cohorts, the toxicities observed were similar to that observed in the continuous dosing cohorts.
Dose interruptions due to AEs were common, occurring in 29 (42 %) of patients due primarily to skin and GI-related toxicities. Thirteen patients (19 %) had at least one AE leading to permanent discontinuation of study treatment. Treatment related dose reductions were reported in 4 % of patients. Sixteen treatment related serious AEs occurred in 8 patients including rash (3), asthenia/fatigue (3), stomatitis (2), pyrexia (2), and one each of dehydration, diarrhea, hypertension, left ventricular dysfunction, acute renal failure, and vomiting. A summary of drug-related AEs by maximum toxicity grade across all dose combinations is shown in Fig. 2. Nine deaths were reporting during the study period, none of which were related to study treatment.

Summary of drug-related adverse events by maximum toxicity grade across all dose combinations. a The category of rash was comprised of the following collapsed preferred terms: dermatitis acneiform, erythema, rash, rash erythematous, rash macular, and rash maculo-papular. b The category of mucositis was comprised of the following collapsed preferred terms: aphthous stomatitis, mouth ulceration, mucosal inflammation, and stomatitis
Pharmacokinetics
GSK458 had an estimated population clearance of 3.25 L/h, inter-compartment clearance of 2.71 L/h, central and peripheral volume of distribution of 8.95 and 38.4 L respectively (Table 4). The inter-individual variability on clearance and central volume was approximately 53 % and 97 % respectively. PK of the GSK458 was compared when administered alone as single agent and in combination with trametinib. No significant difference was observed in post hoc estimates following administration of GSK458.
Efficacy
Of the 69 patients, 57 were assessable for radiographic response (Fig. 3). One partial response was observed in a KRAS mutant ovarian cancer patient who remained on study for >400 days at the time of data analysis in the continuous BID dosing cohort. Stable disease >16 weeks was observed in 12 patients. Of the 48 patients enrolled in the continuous daily dosing cohort, plasma at baseline was available for 30 patients to evaluate for cfDNA. Mutations in KRAS, NRAS, BRAF or PIK3CA were detected in 21/30 (70 %). None of the three patients (ocular melanoma, renal cell carcinoma, and gall bladder cancer) in the continuous daily dosing cohort with stable disease >16 weeks had detectable cfDNA at baseline.

Time on study treatment by disease type by RECIST version 1.1 best response category.* Non-CR/Non-PD (green), Non-complete response/Non-progressive disease; PD (purple), Progressive disease; NE (orange),Not evaluable; SD (red), Stable Disease; PR (blue), partial response. * This figure reflects the updated treatment duration from database lock on 01 May 2014 (last patient off treatment on March 31st, 2015)
Discussion
In this study, we established two MTDs for continuous once daily dosing: 2.0 mg GSK458/1.0 mg trametinib, and 1.0 mg GSK458/1.5 mg trametinib. We determined that 0.5 mg GSK458 BID continuous plus 1.5 mg trametinib QD exceeded the MTD; the MTD was not established for GSK458 intermittent dosing (4 days on, 10 days off) prior to study termination. Even at low doses of GSK458 and trametinib tolerability was poor, primarily due to skin and GI-related toxicities that necessitated dose interruptions in 42 % and treatment discontinuation in 19 %. Exposures to GSK458 in this study were similar to GSK458 monotherapy [13], indicating that our inability to administer GSK458 at doses near the single agent MTD in combination with trametinib was not due to a PK interaction.
Despite enrichment for tumor types with somatic PI3K and/or MAPK pathway alterations, minimal anti-tumor activity was observed with the GSK458 and trametinib combination. This is inconsistent with extensive pre-clinical data showing additive or synergistic responses with the combination [2–4]. Other trials with PI3K or AKT and MEK inhibitor combinations have also reported poor tolerability, with frequent overlapping side effects including rash, diarrhea, nausea, and mucositis, limiting the doses of either drug administered in combination and demonstrating disappointing efficacy [14–20]. In preclinical studies, intermittent dosing regimens of a PI3K and MEK combination showed similar activity to a continuous dosing schedule with improved tolerability [21]. Here, we explored intermittent dosing of GSK458 (4 days on, 10 days off) with continuous daily dosing of trametinib but failed to observe improved tolerability. Further exploration of the intermittent schedule was discontinued before the MTD was defined after modeling predicted that we would be unable to deliver GSK458 and trametinib in combination doses sufficient to produce adequate inhibition of the PI3K/AKT and RAS/RAF/MEK pathways required for anti-tumor activity in preclinical models.
There are several limitations to our study. First, our study was terminated prior to completion of the planned PK studies to assess the effect of GSK458 on trametinib exposures at the MTD. Second, only two intermittent dosing schedules were explored. We did not evaluate an intermittent schedule of trametinib, due to its long plasma half-life of five days. It is possible that intermittent scheduling of another PI3K or AKT and MEK inhibitor combination might be associated with improved tolerability, particularly with a rest period from both drugs. Third, the study was terminated before a planned serial biopsy cohort was enrolled; therefore, we were not able to assess the level of pharmacodynamic inhibition of the PI3K/AKT and RAS/RAF/MEK pathways in tumor tissues at the MTDs defined.
The NCI Investigational Drug Steering Committee has recently published recommendations for phase I with novel drug combinations [22]. Our experience with GSK458 and trametinib highlights the importance of rigorous assessment of DLTs and drug exposures for novel combinations with overlapping toxicities that may limit optimal dosing. Due to poor tolerability and limited anti-tumor activity no further development of the GSK458 and trametinib combination is planned, which is in keeping with termination of other PI3K or AKT and MEK inhibitor combinations [20, 23, 24].
References
McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D’Assoro AB (2006) Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzym Regul 46(1):249–279
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y (2008) Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 14(12):1351–1356
Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, Heynck S, Stückrath I, Weiss J, Fischer F (2009) Identifying genotype-dependent efficacy of single and combined PI3K-and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci 106 (43):18351–18356
Hoeflich KP, O'Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T (2009) In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res 15(14):4649–4664
Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, Bayani N, Wang NJ, Neve RM, Guan Y (2009) Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res 69(2):565–572
Munster P, van der Noll R, Voest E, Specht J, Werner T, Dees E, Tan A, Daud A, Schellens J, Lolkema M (2012) PI3K kinase inhibitor GSK2126458 (GSK458): Clinical activity in select patient (PT) populations defined by predictive markers (study P3K112826). In: Ann Oncol, . Oxford univ press great clarendon ST, Oxford OX2 6DP, England, pp 153–154
Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR (2012) Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol 13(8):773–781
Eisenhauer E, Therasse P, Bogaerts J, Schwartz L, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz Jr LA (2008) Circulating mutant DNA to assess tumor dynamics. Nat Med 14 (9):985–990. http://www.nature.com/nm/journal/v14/n9/suppinfo/nm.1789_S1.html
Gasparini M, Bailey S, Neuenschwander B (2010) Bayesian dose finding in oncology for drug combinations by copula regression. J R Stat Soc: Series C (Applied Statistics) 59(3):543–544
Riviere MK, Yuan Y, Dubois F, Zohar S (2014) A Bayesian dose-finding design for drug combination clinical trials based on the logistic model. Pharm Stat 13(4):247–257. doi:10.1002/pst.1621
Tighiouart M, Rogatko A, Babb JS (2005) Flexible Bayesian methods for cancer phase I clinical trials. Dose escalation with overdose control. Stat Med 24(14):2183–2196. doi:10.1002/sim.2106
Munster P, van der Noll R, Voest E, Dees E, Tan A, Specht J, Falchook G, Daud A, Lolkema M, Grilley-Olson J (2011) Phase I first-in-human study of the PI3 kinase inhibitor GSK2126458 (GSK458) in patients with advanced solid tumors (study P3K112826). In: ASCO annual meeting proceedings, 15_suppl. p 3018
Tolcher A, Baird R, Patnaik A, Moreno Garcia V, Papadopoulos K, Garrett C, Olmos D, Shannon K, Zazulina V, Eubin E (2011) A phase I dose-escalation study of oral MK-2206 (allosteric AKT inhibitor) with oral selumetinib (AZD6244; MEK inhibitor) in patients with advanced or metastatic solid tumors. J Clin Oncol 29:3004
Infante JR, Gandhi L, Shapiro G, Burris HA, Bendell JC, Baselga J, Hsu K, Faivre T, Asatiani E, Heist RS (2012) Phase lb combination trial of a MEK inhibitor, pimasertib (MSC1936369B), and a PI3K/mTOR inhibitor, SAR245409, in patients with locally advanced or metastatic solid tumors. In: J Clin Oncol, vol 15. Amer soc clinical oncology 2318 mill road, STE 800, alexandria, VA 22314 USA,
Bedard P, Tabernero J, Kurzrock R, Britten CD, Stathis A, Manuel Perez-Garcia J, Zubel A, Le NT, Carter K, (2012) Bellew KM A phase lb, open-label, multicenter, dose-escalation study of the oral pan-PI3K inhibitor BKM120 in combination with the oral MEK1/2 inhibitor GSK1120212 in patients (pts) with selected advanced solid tumors. In: J Clin Oncol, vol 15. Amer soc clinical oncology 2318 mill road, STE 800, Alexandria, VA 22314 USA,
Shapiro G, LoRusso P, Kwak E, Cleary J, Musib L, Jones C, de Crespigny A, Belvin M, McKenzie M, Gates M (2011) Clinical combination of the MEK inhibitor GDC-0973 and the PI3K inhibitor GDC-0941: A first-in-human phase Ib study testing daily and intermittent dosing schedules in patients with advanced solid tumors. J Clin Oncol 29(15 suppl):3005
Bendell JC, LoRusso P, Cho DC, Musib L, Yan Y, Chang I, Patel P, Chang IT, Meng RD, Shapiro GI (2014) Clinical results of a phase Ib dose-escalation study of the Mek inhibitor cobimetinib (GDC-0973) and the Akt inhibitor ipatasertib (GDC-0068) in patients (pts) with solid tumors. In: American association for cancer research annual meeting, p CT328
Kurzrock R, Patnaik A, Rosenstein L, Fu S, Papadopoulos K, Smith D, Falchook G, Chambers G, Gauvin J, Naing A (2011) Phase I dose-escalation of the oral MEK1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral AKT inhibitor GSK2141795 (GSK795). J Clin Oncol 29:215 s
Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, Orford K, Aktan G, Ferron-Brady G, Ibrahim N (2015) Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother Pharmacol 75(1):183–189
Hoeflich KP, Merchant M, Orr C, Chan J, Den Otter D, Berry L, Kasman I, Koeppen H, Rice K, Yang N-Y (2012) Intermittent administration of MEK inhibitor GDC-0973 plus PI3K inhibitor GDC-0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res 72(1):210–219
Paller CJ, Bradbury PA, Ivy SP, Seymour L, LoRusso PM, Baker L, Rubinstein L, Huang E, Collyar D, Groshen S (2014) Design of Phase I Combination Trials: Recommendations of the Clinical Trial Design Task Force of the NCI Investigational Drug Steering Committee. Clin Cancer Res 20(16):4210–4217
Bedard PL, Tabernero J, Janku F, Wainberg ZA, Paz-Ares L, Vansteenkiste J, Van Cutsem E, Pérez-García J, Stathis A, Britten CD, Le N, Carter K, Demanse D, Csonka D, Peters M, Zubel A, Nauwelaerts H, Sessa C (2014) A Phase Ib Dose-Escalation Study of the Oral Pan-PI3K Inhibitor Buparlisib (BKM120) in Combination with the Oral MEK1/2 Inhibitor Trametinib (GSK1120212) in Patients with Selected Advanced Solid Tumors. Clin Cancer Res. doi:10.1158/1078-0432.ccr-14-1814
Janku F, Stathis A, Perez-Garcia J, Wainberg Z, Paz-Ares L, Vansteenkiste J, Van Cutsem E, Le N, Zubel A, Bedard P Phase Ib study of oral pan-PI3K BKM120 in combination with the oral MEK1/2 inhibitor GSK120212 in patients with selected advanced solid tumors and RAS/BRAF mutations. In: Eur J Cancer, 2013. Elsevier sci ltd the boulevard, langford lane, Kidlington, Oxford Ox5 1gb, Oxon, England, pp S162-S162
Acknowledgments
The authors would like to thank the patients who participated in the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
This study was funded by GlaxoSmithKline. Rajendra Singh and Yuehui Wu are current employees/stockholders of GlaxoSmithKline. Leanne Cartee is a former employee and current stockholder of GlaxoSmithKline. Philippe Bedard has received research funding from GlaxoSmithKine and Novartis. Albiruni Razak has received travel funding from GlaxoSmithKline, Novartis, BristolMyersSquibb, Boehringer, Karyopharm, and Deciphera. All remaining authors declare no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all participants included in the study.
Additional information
J. E. Grilley-Olson and P. L. Bedard contributed equally to this article.
Highlights
•We report a phase Ib study of the PI3K/mTOR inhibitor, GSK2126458, and trametinib
•The combination is poorly tolerated, due to overlapping skin and GI toxicities
•Efficacy was limited, possibly due to dosing limitations
Rights and permissions
About this article

Cite this article
Grilley-Olson, J.E., Bedard, P.L., Fasolo, A. et al. A phase Ib dose-escalation study of the MEK inhibitor trametinib in combination with the PI3K/mTOR inhibitor GSK2126458 in patients with advanced solid tumors. Invest New Drugs 34, 740–749 (2016). https://doi.org/10.1007/s10637-016-0377-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-016-0377-0