
Summary
Purpose 5-imino-13-deoxydoxorubicin (DIDOX; GPX-150) is a doxorubicin analog modified in two locations to prevent formation of cardiotoxic metabolites and reactive oxygen species. Preclinical studies have demonstrated anti-cancer activity without cardiotoxicity. A phase I study was performed in order to determine the maximum-tolerated dose (MTD) of GPX-150 in patients with metastatic solid tumors. Methods GPX-150 was administered as an intravenous infusion every 21 days for up to 8 cycles. An accelerated dose escalation was used for the first three treatment groups. The dosing groups were (A) 14 mg/m2, (B) 28 mg/m2, (C), 56 mg/m2, (D) 84 mg/m2, (E) 112 mg/m2, (F) 150 mg/m2, (G) 200 mg/m2, and (H) 265 mg/m2. Pharmacokinetic samples were drawn during the first 72 h of cycle 1. Results The MTD was considered to be reached at the highest dosing level of 265 mg/m2 since dose reduction was required in 5 of 6 patients for neutropenia. The most frequent adverse events were neutropenia, anemia, fatigue, and nausea. No patients experienced cardiotoxicity while on the study. The best overall response was stable disease in four (20 %) patients. Pharmacokinetic analysis revealed an AUC of 8.0 (±2.6) μg · h/mL, a clearance of 607 (±210) mL/min/m2 and a t1/2β of 13.8 (±4.6) hours. Conclusions GPX-150 administered every 21 days has an acceptable side effect profile and no cardiotoxicity was observed. Further investigation is needed to determine the efficacy of GPX-150 in anthracycline-sensitive malignancies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anthracyclines, including doxorubicin, daunorubicin, epirubicin, and idarubicin, represent one of the most commonly used class of anti-neoplastic agents. First discovered in the 1950’s, these agents remain the backbone of therapy for a number of solid tumor and hematological malignancies, including, but not limited to, breast cancer, gastric cancer, ovarian cancer, sarcoma, lymphoma, acute myeloid/lymphoblastic leukemia and multiple myeloma. Cardiotoxicity is one of the most significant toxicities of the anthracyclines and limits the cumulative amount of drug able to be administered. Anthracyclines can elicit an early (acute) and delayed (chronic) cardiotoxicity [1, 2]. Acute cardiotoxicity can manifest with ECG changes, dysrhythmias, pericarditis, myocarditis, and decreases in cardiac contractility. Both early and late chronic cardiotoxicity have been recognized. Early chronic toxicity can arise within weeks or months after completion of anthracycline therapy (median 1 month) [1]. Steinherz et al. described a protracted cardiotoxicity occurring 4 to 20 years after completion of therapy (late chronic) [3]. Early studies suggested that approximately 2 % of patients treated with doxorubicin developed signs and symptoms of congestive heart failure [4]. Later studies demonstrated that approximately 5 % of patients had a significant decrease in left ventricular function or evidence of congestive heart failure [5]. Furthermore, the development of congestive heart failure was noted to be associated with the total cumulative dose of doxorubicin [4, 6, 5].
The cytotoxic effects of anthracyclines are related to their ability to intercalate DNA and inhibit DNA replication via topoisomerase II α [7]. In contrast, the cardiotoxic effects of anthracyclines appear to be a consequence of free radical formation that occurs during metabolism. Doxorubicin (Fig. 1) consists of a naphthacenequinone nucleus linked through a glycosidic bond at ring atom 7 to an amino sugar, daunosamine. NADH dehydrogenase [8] and nitric oxide synthase [9] reduce the quinine moiety of doxorubicin to form a semiquinone radical than can redox cycle and reduce oxygen to superoxide and form other reactive oxygen species (ROS). In addition, doxorubicin and daunorubicin are metabolized via the enzyme carbonyl reductase to form doxorubicinol (Fig. 1) and daunorubicinol, respectively. These metabolites interfere with cellular iron regulation yielding ROS and initiating cardiac myocyte apoptosis [10]. Animal studies have demonstrated that the cardiac concentrations of daunorubicinol and doxorubicinol, but not the parent drug concentrations, correlate with cardiac dysfunction [11–13]. Other studies demonstrate that anthracyclines inhibit cardiac function by impairing cardiac calcium regulation through a mechanism dependent upon sarcoplasmic reticulum function and the anthracycline quinone moiety [14]. Anthracycline cardiotoxicity has been linked to poisoning of cardiac topoisomerase IIβ with formation of ROS and activation of cardiac apoptosis [14, 15].

Chemical structures of doxorubicin, doxorubicinol, GPX-100, and GPX-150
Approaches to the development of noncardiotoxic anthracyclines have included aldoxorubicin and AD-198. Aldoxorubicin, a 6-maleimidocaproyl hydrazone derivative of doxorubicin, is a pro-drug which covalently binds to albumin once in the bloodstream, and is cleaved inside cells [16–19]. This agent is thought to have less cardiotoxicity than doxorubicin because of lower drug accumulation in organs and lower plasma doxorubicinol concentrations [16–19]. Aldoxorubicin has been tested in phase I and II clinical trials with no evidence of acute cardiotoxicity, although myelosuppression and mucositis were noted [19–21]. AD 198 (N-benzyladriamycin-14-valerate) is a doxorubicin derivative which retains the semiquinone structure but has less cardiotoxicity in the pre-clinical setting, possibly as a consequence of increased protein kinase C epsilon activity [22]. This agent has not been tested in clinical trials. The risk of cardiotoxicity decreases with a liposomal formulation of doxorubicin (Doxil), but is still present [23].
As an alternative approach, two derivatives of doxorubicin, GPX-100 and GPX-150 (Fig. 1), have been developed as potentially non-cardiotoxic agents. GPX-100 (13-deoxydoxorubicin) lacks the carbonyl group at the C-13 carbon which in the parent compound is reduced to form doxorubicinol. This compound has previously been shown to be a very poor substrate for carbonyl reductase [24]. GPX-150 (13-deoxy-5-iminodoxorubicin) also lacks the C-13 carbonyl group and the quinone moiety has been converted to an iminoquinone. A Phase I study of GPX-100 in 16 patients with solid tumors determined the MTD to be 140 mg/m2 [25]. A subsequent small Phase 2 study involving seven patients with metastatic breast cancer was stopped early because of evidence of cardiotoxicity (decreased left ventricular ejection fraction and electromechanical dissociation) implicating the importance of the quinone moiety in anthracycline-induced cardiotoxicity.
As GPX-150 lacks both the C13 carbonyl and the quinone moieties, it was hypothesized that this agent would retain cytotoxic activity yet be noncardiotoxic. This phase I dose escalation study of GPX-150 in patients with advanced malignancy further supports this hypothesis, as at doses as high as 265 mg/m2, no acute cardiotoxicity was observed.
Materials and methods
The trial was conducted at the University of Iowa and University of Texas at San Antonio and was approved by the corresponding Institutional Review Boards. Informed consent was obtained from all individual participants included in the study. The trial was registered with ClinicalTrials.gov (Identifier No. NCT00710125).
Objectives
The primary objectives were to determine the maximum tolerated dose (MTD) and dose-limiting toxicity (DLT) in patients with solid tumors. The secondary objectives were to evaluate the pharmacokinetics and metabolism of GPX-150 and to document any anti-tumor activity.
Eligibility
Individuals (age ≥ 18 years) with advanced metastatic cancer with progressive disease for whom no effective standard therapy was available, and who gave informed written consent according to Food and Drug Administration and institutional guidelines, were eligible. Patients had to have a performance status of at least 70 % on the Karnofsky scale. Patients must not have received any cytotoxic chemotherapy, hormonal therapy, other investigational agents, or palliative radiation within 4 weeks of the first treatment on study. Patients were required to have adequate bone marrow function (ANC count ≥1500/mm3, hemoglobin ≥ 9.0 g/dL, and platelet count ≥100,000/mm3), renal function (serum creatinine ≤2.0 mg/dl or estimated creatinine clearance ≥ 50 ml/min), and hepatic function (bilirubin ≤1.5 × ULN, AST and ALT < 2.5 × ULN (<5.0 × ULN with liver involvement)). Patients were required to have an ejection fraction of 110 % of the lower limit of the institutional normal as determined by resting MUGA and have a baseline oxygen saturation by pulse oximetry of at least 90 %. Exclusion criteria included: pregnancy or breast feeding; history of hypersensitivity to anthracyclines; having received a cumulative dose of doxorubicin exceeding 300 mg/m2 or a cumulative dose of epirubicin exceeding 450 mg/m2; having received an anthracycline within 6 months prior to study entry; brain metastases unless asymptomatic and stable off glucocorticoids; prior history of congestive heart failure (CHF); myocardial infarction within 6 months prior to enrollment, active ischemic heart disease, or uncontrolled hypertension; requiring active medical therapy for CHF or arrhythmia; > grade 1 motor neuropathy or > grade 2 sensory neuropathy; or lymphoma.
Drug Formulation and Administration
GPX-150 was supplied (by Gem Pharmaceuticals, LLC, Birmingham, AL) as a lyophilized powder formulation in sterile 25 mL amber glass vials containing 36 mg of drug and 250 mg of lactose monohydrate, USP. Vials were protected from light and stored at -20 °C. The lyophilized powder was reconstituted by the addition of 25 mL of 0.9 % sodium chloride injection, USP, to the vial. The drug was administered intravenously at a rate of 2 mL/min.
Dose Escalation
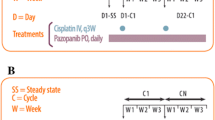
The study was designed so that the first three treatment groups had a minimum of one patient per group and there was an increase in an accelerated fashion of 100 % over the previous dose. If a grade 2 or higher toxicity was observed during the accelerated dose titration, then 2 additional patients would be added to that and subsequent dose levels. The fourth dose group was a 50 % increase over the previous group. Doses were escalated by 33 % after the fourth cohort. After the first three dose groups, all subsequent groups were to have a minimum of three patients per group until the last cohort which would have six patients. If two of three patients in a group experienced a dose limiting toxicity (DLT), then six patients total would be enrolled in that cohort. Dose reductions of 25 % were required for a ≥ grade 2 non-hematological toxicity or a ≥ grade 3 hematological toxicity. The dosing groups were as follows: (A) 14 mg/m2, (B) 28 mg/m2, (C) 56 mg/m2, (D) 84 mg/m2, (E) 112 mg/m2, (F) 150 mg/m2, (G) 200 mg/m2 and (H) 265 mg/m2. Patients who required dose reduction could not have their dose re-escalated.
Dosing schedule
The dosing schedule for all groups was once every 3 weeks. Treatment could be continued for up to 8 cycles in anthracycline-naïve patients and up to 4 cycles in patients previously treated with anthracyclines. Prior to each scheduled dose, the ANC must have recovered to ≥ 1500 cells/mL, the platelet count to ≥ 100,000/mL and all acute non-hematologic toxicity to ≤ grade 1 or baseline. If the parameters were not met, patients were evaluated weekly and treated when the parameters were met. However, if there were delays greater than 2 weeks, patients would be removed from the study.
Assessment of Safety
The pretreatment evaluation included physical examination, documentation of all sites of disease with baseline radiologic evaluations, and assessment of performance using the Karnofsky scale. Patients also had an ECG, a MUGA scan, standard PFTs with oxygen saturation by pulse oximetry, and laboratory studies. Study treatment visits were at 3-week intervals at which time the patients underwent physical exam. Laboratory studies consisting of a complete blood count (CBC) with differential and chemistry screen were obtained on days 8 and 15 of each cycle. In addition, a CBC with differential was obtained on day 11 or 12 of each cycle in order to determine the hematological nadir. PFT and oxygen saturation were obtained prior to cycles 3, 5, and 7 and at 4 weeks following completion of the last cycle. Toxicities were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, v3.0. A DLT was defined as fever with grade 4 neutropenia, grade 4 neutropenia lasting ≥ 5 days, grade 4 thrombocytopenia, inability to start the next cycle after a 2-week delay, grade 3 or 4 vomiting with maximum supportive care, ≥ grade 2 neurotoxicity, ≥ grade 2 decrease in cardiac ejection fraction, ≥ grade 3 cardiac rhythm or conduction abnormalities, or any other toxicity ≥ grade 3 excluding nausea or alopecia. The MTD was defined as the maximum tolerated dose level at which no more than two instances of DLT were observed in up to 6 patients.
Assessment of cardiac function
ECG and MUGA scans were obtained prior to cycles 3, 5, and 7 as well as 4 weeks following completion of the last cycle. For patients with prior treatment with anthracyclines, assessment of cardiac function was performed before every cycle. In the event of grade 2 cardiotoxicity, patients would be monitored with ECG and MUGA scan every 6–8 weeks until cardiac function stabilized or improved and further GPX-150 would not be administered.
Assessment of response
Imaging studies were performed at baseline and after every two subsequent cycles, or at discontinuation from study. The RECIST criteria were used to determine response.
Pharmacokinetic studies
Samples for pharmacokinetic studies collected during the first cycle. Blood samples were drawn at baseline, 15, 30, and 45 min after beginning infusion and then at 1, 1.5, 2, 4, 6, 8, 24, 48, and 72 h after the end of the infusion period. In addition, timed urine collections were obtained (0–12, 12–24, 24–48, 48–72 h). GPX-150 in human plasma and urine was measured using a method developed by Microconstants, San Diego, CA. Plasma and urine samples containing GPX-150, an ethyl chloroformate derivatized form of GPX-150 as the internal standard, and lithium heparin as the anticoagulant, were processed by a method involving protein precipitation, derivatization, and liquid/liquid extraction. During the extraction, the standards and samples were derivatized with propyl chloroformate. An aliquot of the extract was analyzed by high-performance liquid chromatography using a Develosil RP Aqueous column. The mobile phase was nebulized using heated nitrogen in a Z-spray source/interface and the ionized compounds were detected using a tandem quadrupole mass spectrometer. GPX-150 was derivatized and separated from plasma and urine by HPLC (ACQUITY, Waters) using a 150 × 2.0 mm, 5 μm Develosil RP Aqueous column (Phenomenex) and MS/MS (Quattro Premier, Micromass). HPLC mobile phase was composed of two solutions (37%A and 63%B) of the following composition: Mobile Phase A: 10 % Ammonium Formate:1.25 % Citric Acid in water:Formic Acid:Water (3.2:0.173:1.6:795, v/v/v/v) and Mobile Phase B: 10 % Ammonium Formate:1.25 % Citric Acid in water:Formic Acid:Water:Acetonitrile (3.2:0.173:1.6:19.2:776, v/v/v/v/v). Peak areas of GPX-150 and the internal standard were obtained using MassLynx v. 4.1 software (Waters, Milford, MA). The calibration curves were obtained by fitting the peak area ratios of GPX-150/internal standard and the standard concentrations to a ln-transformed linear equation using MassLynx. The equations of the calibration curves were then used to interpolate the concentrations of GPX-150 in the samples using their peak area ratios.
The PK parameters were determined via non-compartmental analysis by applying a non-linear curve fit using the Levenberg-Marquardt fitting method and commercial software (XLFit, IDBS London, UK) to the post-infusion decay data. This PK analysis was performed on the data obtained from the seven patients in dose level H. Summation of the coefficients for each subject yielded a range of estimated maximum plasma concentration (Cmax) at the time of discontinuation of the infusions. The terminal half-life (t1/2β) was obtained from the lowest exponents of the fit. A bi-exponential model using the Wagner correction was employed to determine the systemic clearance (Cl), the area under the plasma concentration-time curve from zero to infinity (AUC0-∞), and the volume of distribution at steady-state (Vdss) [26, 27].
[3H]-Thymidine incorporation assay
HL-60 cells were obtained from the American Type Culture Collection (ATCC) (Manassas, VA) and cultured in RPMI-1640 media supplemented with 10 % fetal bovine serum and penicillin/streptomycin at 37 °C and 5 % CO2. Cells (15,000 cells/100 μL/well) were incubated in 96-well plates in the presence or absence of varying concentrations of doxorubicin (25 nM-2 μM) or GPX-150 (75 nM-6 μM) for 24 h. Tritiated thymidine (ICN) (1 μCi/50 μL) was then added to each well. Following a 4-h incubation, cells were lysed with deionized water and cellular contents collected onto Whatman GF-C filters using a Brandel cell harvester. Filters were dried and counted using liquid scintillation counting. The results were analyzed by curve fitting analysis using nonlinear regression (PRISM, GraphPad software, San Diego, CA).
Results
Anti-proliferative activity of GPX-150
[3H]-thymidine incorporation assays performed in HL60 cells demonstrated that both GPX-150 and doxorubicin produce concentration-dependent decreases in proliferation (Fig. 2). Doxorubicin was noted to be four-fold as potent as GPX-150 in inhibiting thymidine incorporation (IC50 148 nM for doxorubicin vs 593 nM for GPX-150).

Anti-proliferative effects of GPX-150 and doxorubicin. [3H]-thymidine incorporation assays were performed using HL-60 cells incubated in the presence or absence of GPX-150 or doxorubicin for 24 h. Values are expressed as percentage of untreated control (mean ± SEM) of three separate experiments replicated six times per experiment
Patient Demographics
A total of 24 patients were enrolled in this study from December 2007 to April 2013. The trial was closed for accrual for a period between 2009 and 2011 because of changes in sponsor. All patients received at least one dose of study drug and were included in the safety analysis. The characteristics of the subjects are shown in Table 1. Sarcomas were the most common malignancy. The majority of the subjects had received multiple lines of prior therapy. Nearly half of the subjects had previously received radiation therapy. A total of 4 patients had received prior anthracycline therapy.
Dose Escalation and Toxicity
As shown in Table 2, dose escalation proceeded as planned with the exception that one additional patient each was enrolled in dose levels D, G, and H to make up for the early termination of three patients (one per dose level). The most frequent AE’s were hematologic (anemia and neutropenia), fatigue, and nausea (Table 3). Of note, there were no episodes of febrile neutropenia. There were no clinically significant changes in cardiac function in any patients, including in patients with prior anthracycline exposure, and no left ventricular ejection fractions dropped below 50 % (Table 4). A DLT was not observed and the MTD was not formally reached at the highest dose level. Of the six patients at the highest dose level who received more than one cycle, all but one required dose reduction because of neutropenia and/or anemia and in total, 60 % of the cycles were dose-reduced. Thus the 265 mg/m2 dose level was the highest dose tolerated in the study. The one patient who completed 4 cycles at 265 mg/m2 before progressing had metastatic renal cell cancer previously treated with non-cytotoxic agents (sunitinib, everolimus, and IL-2).
Pharmacokinetics
Table 5 presents the pharmacokinetic data for GPX-150 following the first dose on study at the 265 mg/m2 dosing level. Although the study protocol dictated that blood samples be drawn at various time points following the completion of study drug infusion, in actuality, the 15 min, 30 min, and 45 min samples were collected after the beginning of the infusion. The one hour and subsequent time points were collected following the completion of the infusion. A bi-exponential model with the Wagner correction method was used to determine the PK parameters based on the post-infusion data [26, 27]. Excellent bi-exponential fits were obtained for all data sets with no R2 value being less than 0.995 (0.997 ± 0.002). As shown in Fig. 3, GPX-150 in plasma was eliminated quickly. Measurement of GPX-150 in the urine of patients following their first dose revealed that approximately 5–10 % of the total administered dose was excreted in the urine, primarily within the first 24 h (Fig. 4). These measurements are consistent with the patients’ observations that their urine had a purple color during the first day following treatment.

Semi-log plot of plasma GPX-150 concentration versus time in three representative patients in dose level H (265 mg/m2) following first dose administration

Urinary excretion of GPX-150 following first dose administration in three representative patients in dose level H (265 mg/m2)
Response to Therapy
Of the 20 patients who were evaluable for response by virtue of receiving at least two cycles of therapy, four had stable disease at the conclusion of the study and the remainder progressed while on study (Supplementary Table 1). Two additional patients, one in dose level E and one in dose level H, completed 8 cycles but had evidence of disease progression at the conclusion of the study. The four patients who achieved stable disease included a desmoid (dose level E, three prior lines of therapy), an adenoid cystic carcinoma (dose level F, two prior lines of therapy), a uterine leiomyosarcoma (dose level F, one prior line of therapy), and a urethral carcinoma (dose level H, no prior lines of therapy).
Discussion
This first-in-human study of the novel doxorubicin analogue GPX-150 in patients with advanced solid tumors reveals that this agent may be safely administered at doses as high as 265 mg/m2. Although a formal MTD was not reached, 5 of 6 patients at the highest dose level required a dose-reduction because of neutropenia. Thus we considered 265 mg/m2 the highest dose achievable without the use of granulocyte colony stimulating factor (G-CSF) drugs. Importantly, no cardiotoxicity was observed during this study, even in patients with prior anthracycline exposure. The design of this phase I study did not afford opportunity to assess for long term changes in cardiac function. While larger studies are needed to confirm the lack of cardiotoxicity, these phase I results are highly encouraging.
There have been multiple studies which have evaluated the pharmacokinetics of doxorubicin [28–33]. These studies have demonstrated the t1/2 of doxorubicin to be in the 16–40 h range and clearance to be in the 412–677 mL/min/m2 range, similar to GPX-150 (Table 5). As with doxorubicin, the GPX-150 pharmacokinetic data are consistent with bi-exponential kinetics. The amount of unchanged drug excreted in the urine (Fig. 4) is similar to that observed for doxorubicin [32], consistent with primarily fecal excretion and a large VD. Thus it does not appear that the chemical modifications to doxorubicin which yielded GPX-150 result in significant alterations in the half-life or clearance.
Although grades 3–4 neutropenia were observed at the highest dose level, there were neither episodes of febrile neutropenia nor delays in initiating subsequent cycles. Nevertheless, for safety, dose reduction was required per protocol in 5 of the 6 patients at the 265 mg/m2 dose level. Thus it is reasonable to predict that even higher doses of GPX-150 could be safely administered if used in combination with G-CSF support such as is commonly done with anthracycline-containing regimens [34, 35]. As pre-clinical studies have demonstrated that doxorubicin is approximately four times more potent than GPX-150 (Fig. 2), it can be estimated that the 265 mg/m2 dose of GPX-150 is equivalent to approximately 65 mg/m2 of doxorubicin, within the dosing range most commonly used for doxorubicin.
Unlike other anthracyclines, including doxorubicin, liposomal doxorubicin (Doxil), and aldoxorubicin [21, 20], mucositis, stomatitis, or hand-foot syndrome did not occur in any patient treated with GPX-150. With such a favorable side effect profile, and in the absence of detectable cardiotoxicity, GPX-150 may be able to be administered for more than 8 cycles without limitation to the total cumulative dose. Additional studies with longer term follow-up are required to demonstrate the lack of late chronic cardiotoxicity. However, if these studies were to demonstrate the absence of late chronic cardiotoxicity, then GPX-150 would be the only anthracycline without cumulative dose limitations and thus would allow for the generation of treatment regimens that increase the duration of disease control in patients who respond to GPX-150. This would be particularly significant in the setting of metastatic sarcoma, for example, where there are limited treatment options and for which doxorubicin remains one of the most active agents.
The favorable side effect profile observed with GPX-150 also supports the hypothesis that it could be used in combination with other chemotherapy agents without significantly increasing the toxicity profile. Anthracyclines remain an important class of drug in both the adjuvant and metastatic breast cancer setting, but because of trastuzumab’s cardiotoxic effects, combinations with anthracyclines and trastuzumab are generally avoided in HER2- positive patients. Whether GPX-150 could be used in place of traditional anthracyclines in regimens such as CHOP for aggressive lymphomas, ABVD for Hodgkin lymphoma, Hyper-CVAD for acute lymphoblastic leukemia, or ECF for esophageal/gastric carcinoma remains to be determined, but if this agent has activity in these malignancies, its incorporation into these regimens could result in an improvement in their toxicity profile.
In conclusion, the novel anthracycline derivative GPX-150 was well-tolerated at doses as high as 265 mg/m2 and no changes in cardiac function were observed during treatment. Based on the present study, a phase II study of 265 mg/m2 of GPX-150 in patients with advanced and/or metastatic malignant soft tissue sarcoma is underway (NCT02267083).
References
Bristow MR, Thompson PD, Martin RP, Mason JW, Billingham ME, Harrison DC (1978) Early anthracycline cardiotoxicity. Am J Med 65(5):823–832
Buzdar AU, Marcus C, Smith TL, Blumenschein GR (1985) Early and delayed clinical cardiotoxicity of doxorubicin. Cancer 55(12):2761–2765
Steinherz LJ, Steinherz PG, Tan CT, Heller G, Murphy ML (1991) Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA 266(12):1672–1677
Von Hoff DD, Layard MW, Basa P, Davis HL Jr, Von Hoff AL, Rozencweig M, Muggia FM (1979) Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med 91(5):710–717
Swain SM, Whaley FS, Ewer MS (2003) Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 97(11):2869–2879. doi:10.1002/cncr.11407
Alexander J, Dainiak N, Berger HJ, Goldman L, Johnstone D, Reduto L, Duffy T, Schwartz P, Gottschalk A, Zaret BL (1979) Serial assessment of doxorubicin cardiotoxicity with quantitative radionuclide angiocardiography. N Engl J Med 300(6):278–283. doi:10.1056/NEJM197902083000603
Romero A, Caldes T, Diaz-Rubio E, Martin M (2012) Topoisomerase 2 alpha: a real predictor of anthracycline efficacy? Clin Transl Oncol 14(3):163–168. doi:10.1007/s12094-012-0779-1
Davies KJ, Doroshow JH (1986) Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 261(7):3060–3067
Vasquez-Vivar J, Martasek P, Hogg N, Masters BS, Pritchard KA Jr, Kalyanaraman B (1997) Endothelial nitric oxide synthase-dependent superoxide generation from adriamycin. Biochemistry 36(38):11293–11297. doi:10.1021/bi971475e
Minotti G, Recalcati S, Mordente A, Liberi G, Calafiore AM, Mancuso C, Preziosi P, Cairo G (1998) The secondary alcohol metabolite of doxorubicin irreversibly inactivates aconitase/iron regulatory protein-1 in cytosolic fractions from human myocardium. FASEB J 12(7):541–552
Cusack BJ, Mushlin PS, Voulelis LD, Li X, Boucek RJ Jr, Olson RD (1993) Daunorubicin-induced cardiac injury in the rabbit: a role for daunorubicinol? Toxicol Appl Pharmacol 118(2):177–185
Olson RD, Mushlin PS, Brenner DE, Fleischer S, Cusack BJ, Chang BK, Boucek RJ Jr (1988) Doxorubicin cardiotoxicity may be caused by its metabolite, doxorubicinol. Proc Natl Acad Sci U S A 85(10):3585–3589
Mushlin PS, Cusack BJ, Boucek RJ Jr, Andrejuk T, Li X, Olson RD (1993) Time-related increases in cardiac concentrations of doxorubicinol could interact with doxorubicin to depress myocardial contractile function. Br J Pharmacol 110(3):975–982
Shadle SE, Bammel BP, Cusack BJ, Knighton RA, Olson SJ, Mushlin PS, Olson RD (2000) Daunorubicin cardiotoxicity: evidence for the importance of the quinone moiety in a free-radical-independent mechanism. Biochem Pharmacol 60(10):1435–1444
Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y, Liu LF (2007) Topoisomerase IIbeta mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 67(18):8839–8846. doi:10.1158/0008-5472.CAN-07-1649
Kratz F, Ehling G, Kauffmann HM, Unger C (2007) Acute and repeat-dose toxicity studies of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin (DOXO-EMCH), an albumin-binding prodrug of the anticancer agent doxorubicin. Hum Exp Toxicol 26(1):19–35
Kratz F, Warnecke A, Scheuermann K, Stockmar C, Schwab J, Lazar P, Druckes P, Esser N, Drevs J, Rognan D, Bissantz C, Hinderling C, Folkers G, Fichtner I, Unger C (2002) Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives. Improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J Med Chem 45(25):5523–5533
Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Kratz F, Walker UA (2007) The 6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH) is superior to free doxorubicin with respect to cardiotoxicity and mitochondrial damage. Int J Cancer 120(4):927–934. doi:10.1002/ijc.22409
Mita MM, Natale RB, Wolin EM, Laabs B, Dinh H, Wieland S, Levitt DJ, Mita AC (2014) Pharmacokinetic study of aldoxorubicin in patients with solid tumors. Invest New Drugs. doi:10.1007/s10637-014-0183-5
Chawla SP, Chua VS, Hendifar AF, Quon DV, Soman N, Sankhala KK, Wieland DS, Levitt DJ (2014) A phase 1B/2 study of aldoxorubicin in patients with soft tissue sarcoma. Cancer. doi:10.1002/cncr.29081
Unger C, Haring B, Medinger M, Drevs J, Steinbild S, Kratz F, Mross K (2007) Phase I and pharmacokinetic study of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin. Clin Cancer Res 13(16):4858–4866. doi:10.1158/1078-0432.CCR-06-2776
Hofmann PA, Israel M, Koseki Y, Laskin J, Gray J, Janik A, Sweatman TW, Lothstein L (2007) N-Benzyladriamycin-14-valerate (AD 198): a non-cardiotoxic anthracycline that is cardioprotective through PKC-epsilon activation. J Pharmacol Exp Ther 323(2):658–664. doi:10.1124/jpet.107.126110
Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, Jones A (2010) Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer 10:337. doi:10.1186/1471-2407-10-337
Slupe A, Williams B, Larson C, Lee LM, Primbs T, Bruesch AJ, Bjorklund C, Warner DL, Peloquin J, Shadle SE, Gambliel HA, Cusack BJ, Olson RD, Charlier HA Jr (2005) Reduction of 13-deoxydoxorubicin and daunorubicinol anthraquinones by human carbonyl reductase. Cardiovasc Toxicol 5(4):365–376
Busby L, Fitch T, Perez E, Farmer R, Roedig B, Dechow F, Wheeler RH (2001) A Phase I study of 13-deoxydoxorubicin (GPX-100) using accelerated dose titration. Proc Am Assos Cancer Res 42:834
Wagner JG (1976) Linear pharmacokinetic equations allowing direct calculation of many needed pharmacokinetic parameters from the coefficients and exponents of polyexponential equations which have been fitted to the data. J Pharmacokinet Biopharm 4(5):443–467
Wagner JG (1977) Pharmacokinetic data. Pharmacokinetic parameters estimated from intravenous data by uniform methods and some of their uses. J Pharmacokinet Biopharm 5(2):161–182
Hochster H, Liebes L, Wadler S, Oratz R, Wernz JC, Meyers M, Green M, Blum RH, Speyer JL (1992) Pharmacokinetics of the cardioprotector ADR-529 (ICRF-187) in escalating doses combined with fixed-dose doxorubicin. J Natl Cancer Inst 84(22):1725–1730
Robert J, Bui NB, Vrignaud P (1987) Pharmacokinetics of doxorubicin in sarcoma patients. Eur J Clin Pharmacol 31(6):695–699
Piscitelli SC, Rodvold KA, Rushing DA, Tewksbury DA (1993) Pharmacokinetics and pharmacodynamics of doxorubicin in patients with small cell lung cancer. Clin Pharmacol Ther 53(5):555–561
Rodvold KA, Rushing DA, Tewksbury DA (1988) Doxorubicin clearance in the obese. J Clin Oncol 6(8):1321–1327
Speth PA, van Hoesel QG, Haanen C (1988) Clinical pharmacokinetics of doxorubicin. Clin Pharmacokinet 15(1):15–31. doi:10.2165/00003088-198815010-00002
Ryu RJ, Eyal S, Kaplan HG, Akbarzadeh A, Hays K, Puhl K, Easterling TR, Berg SL, Scorsone KA, Feldman EM, Umans JG, Miodovnik M, Hebert MF (2014) Pharmacokinetics of doxorubicin in pregnant women. Cancer Chemother Pharmacol 73(4):789–797. doi:10.1007/s00280-014-2406-z
Smith TJ, Khatcheressian J, Lyman GH, Ozer H, Armitage JO, Balducci L, Bennett CL, Cantor SB, Crawford J, Cross SJ, Demetri G, Desch CE, Pizzo PA, Schiffer CA, Schwartzberg L, Somerfield MR, Somlo G, Wade JC, Wade JL, Winn RJ, Wozniak AJ, Wolff AC (2006) 2006 update of recommendations for the use of white blood cell growth factors: an evidence-based clinical practice guideline. J Clin Oncol 24(19):3187–3205. doi:10.1200/JCO.2006.06.4451
Aapro MS, Cameron DA, Pettengell R, Bohlius J, Crawford J, Ellis M, Kearney N, Lyman GH, Tjan-Heijnen VC, Walewski J, Weber DC, Zielinski C, European Organisation for R, Treatment of Cancer Granulocyte Colony-Stimulating Factor Guidelines Working P (2006) EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphomas and solid tumours. Eur J Cancer 42(15):2433–2453. doi:10.1016/j.ejca.2006.05.002
Funding/Support
This study was sponsored by Gem Pharmaceuticals, LLC, Birmingham, AL and Coronado Biosciences, Inc., Burlington, MA.
Author’s disclosures of potential conflicts of interest
Sarah A. Holstein: none
James C. Bigelow: none
Richard D. Olson: consultant for Gem Pharmaceuticals, LLC
Robert E. Vestal: consultant for Gem Pharmaceuticals, LLC
Gerald M. Walsh: employee of Gem Pharmaceuticals, LLC
Raymond J. Hohl: research funding from Gem Pharmaceuticals, LLC
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 39 kb)
Rights and permissions
About this article

Cite this article
Holstein, S.A., Bigelow, J.C., Olson, R.D. et al. Phase I and pharmacokinetic study of the novel anthracycline derivative 5-imino-13-deoxydoxorubicin (GPX-150) in patients with advanced solid tumors. Invest New Drugs 33, 594–602 (2015). https://doi.org/10.1007/s10637-015-0220-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-015-0220-z