
Abstract
Background
Hepatic stellate cell hyperactivation is a central link in liver fibrosis development, transforming growth factor β1 (TGF-β1) is a key activator of HSCs.
Aims
This study investigated whether anlotinib attenuates CCl4 induced liver fibrosis in mice and explored its antifibrotic mechanism.
Methods
We used the human hepatic stellate cell line LX-2 for in vitro assays and used TGF-β1 to induce hepatic fibrosis in LX-2 cells. We analyzed cytotoxicity using a cell-counting kit-8 and transwell chambers to detect the migratory ability of LX-2 cells. Western blotting was used to detect the protein levels of collagen type I, α-smooth muscle actin, and p-Smad3. In addition, mice with CCl4-induced hepatic fibrosis were used as in vivo models. Histopathological examination was performed using H&E staining, Masson’s trichrome staining, and immunohistochemistry.
Results
Anlotinib significantly reversed TGF-β1-induced protein levels of Col I, α-SMA and p-Smad3 and inhibits migratory and proliferative abilities in vitro using LX-2 cells. CCl4 cause F4 grade (Ishak) hepatic fibrosis, liver inflammatory scores ranged from 12 to 14 (Ishak), a mean ALT measurement of 130 U/L and a mean measurement AST value of 119 U/L in mice. However, the CCl4-induced changes were markedly attenuated by anlotinib treatment, which returned to F2 grade (Ishak) hepatic fibrosis, liver inflammatory scores ranged from 4 to 6 (Ishak), a mean ALT measurement of 40 U/L and a mean measurement AST value of 56 U/L in mice.
Conclusions
Our results suggest that anlotinib-mediated suppression of liver fibrosis is related to the inhibition of TGF-β1 signaling pathway.
Graphical Abstract
Hepatic stellate cell hyper activation is a central link in liver fibrosis development, transforming growth factor β1 is a key activator of HSCs. Anlotinib is a multi-targeted tyrosine kinase inhibitor that has similar targets to nintedanib, a clinically used anti-pulmonary fibrosis drug. Our study demonstrates an FDA-approved drug—anlotinib—that could prevent liver fibrosis and inflammation. Experiments in cell cultures and mice show that anlotinib can inhibit the activation of hepatic stellate cells by down-regulating the TGFβ1/smad3 pathway, thereby reversing liver fibrosis. In animal experiments, anlotinib showed protective effects on the CCl4-induced liver damage, including ameliorating liver inflammation, reversing liver fibrosis and reducing liver enzymes. This is a very good signal, anlotinib may be useful for halting or reversing the progression of liver fibrosis and could be employed in the development of novel therapeutic drugs for the management of chronic liver diseases.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatic fibrosis is a pathological condition resulting from chronic liver inflammation caused by various factors, including viral hepatitis, alcoholic liver disease, nonalcoholic fatty liver disease, drugs, cholestasis, and autoimmune liver disease, among others [7, 22, 37]. Chronic liver injury leads to impaired liver repair, resulting in liver fibrosis [5]. Continuous activation of hepatic stellate cells (HSCs) and excessive deposition of the extracellular matrix (ECM) caused by liver inflammation are crucial mechanisms in the development of liver fibrosis, and transforming growth factor β1 (TGF-β1) is a key activator of HSCs [5, 9]. During this process, markers of activated HSCs such as α-smooth muscle actin (α-SMA), an important indicator of ECM deposition in the hepatic mesenchyme, and collagen type I (Col I) are upregulated [26]. The inhibition of HSCs has been shown to decrease ECM deposition and alleviate liver fibrosis [4, 32, 36]. Prolonged exposure to these factors increases the degree of liver fibrosis, leading to cirrhosis and liver cancer and resulting in various end-stage liver diseases and high mortality rates [2]. Although cirrhosis and liver cancer are irreversible, early liver fibrosis can be reversed [3, 35]. Unfortunately, the current treatment of liver fibrosis lacks specificity and sensitivity, highlighting the need for further research into the treatment of liver fibrosis.
Anlotinib hydrochloride is a multitarget small-molecule tyrosine kinase inhibitor with antitumor effects in various cancer types, including non-small-cell lung cancer, endometrial cancer, osteosarcoma, and hepatocellular carcinoma [8, 12, 24, 25]. Anlotinib hydrochloride targets receptor tyrosine kinases, including the vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGF), fibroblast growth factor receptor (FGFR), and the non-receptor tyrosine kinase Src [23, 24, 30, 31]. These targets are similar to those of nintedanib, a small-molecule tyrosine kinase inhibitor approved for the treatment of idiopathic pulmonary fibrosis (IPF) [15, 21, 29]. Moreover, recent studies have reported the therapeutic effect of anlotinib on IPF by inhibiting the TGF-β1/Smad signaling pathway and glycolysis pathway [6, 20]. Additionally, anlotinib has shown improvement in live fibrosis and hepatocyte apoptosis induced by bile duct ligation (BDL) by inhibiting the VEGFR2/mTOR pathway [11], and the treatment was proved non-toxic to liver in mice by liver histopathology [34]. Based on its pharmacological action and reported effects on pulmonary and liver fibrosis, we hypothesized that anlotinib could have a therapeutic effect on hepatic fibrosis by inhibiting the TGF-β1/Smad signaling pathway.
The results of this study suggest that anlotinib can reduce carbon tetrachloride (CCl4)-induced liver fibrosis and inhibit excessive ECM deposition by inhibiting the TGF-β1 signaling pathway. Therefore, this study provides a theoretical basis for the clinical development and application of anlotinib in the treatment of hepatic fibrosis.
Materials and Methods
Reagents and Antibodies
Transforming growth factor-β1 (TGF-β1) and CCl4 were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Anlotinib, were purchased from Beijing Solarbio Science & Technology Co., Ltd. The following antibodies were purchased: phospho-smad3 [Ser423/425], Smad3, Anti-collagen I, anti-α-SMA, GAPDH, all obtained from Cell Signaling Technologies (CST). Commercial kits used to measure ALT, and AST were all purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All other chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) and Fisher Scientific (Waltham, MA).
Generation of a Mouse Model of Liver Fibrosis
All animal interventions were approved by the Institutional Animal Care and Use Committee (IACUC) of Guizhou Medical University. The methods and experiment procedures were carried out in accordance with the relevant guidelines and regulations. Mice (C57BL6, 8–10 weeks old) were housed in standard conditions, and sex matched mice were treated with 2.0 µL/g body weight carbon tetrachloride (CCl4) [1:10 (v/v) dilution with corn oil] or corn oil as control by intraperitoneal (i.p.) injection, 3 times per week for 4 weeks. Mice were challenged with CCl4 or corn oil (Control), followed by anlotinib treated (oral, 3 mg/kg/day for 2 week) or 0.9% saline (Vehicle). Mice were sacrificed at 48 h after the last CCl4 injection and tissues were harvested.
Western Blot Analysis
Liver tissues or cells were lysed with RIPA buffer containing a protease inhibitor cocktail. Lysates were centrifuged at 10,000 g for 10 min at 4 °C. The supernatant was collected for further analysis. Total protein was measured using BCA assay kit (Pierce, Carlsbad, CA, USA). Equal amounts of total protein were loaded onto a 10% SDS-PAGE gel and transferred onto polyvinylidene fluoride (PVDF) membranes using a wet transfer device. After blocking with 5% non-fat milk at room temperature for 1 h, membranes were incubated with primary antibodies overnight at 4 °C. Membranes were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. PVDF membranes were incubated with chemiluminescence reagent in order to visualize bands. Collagen I, α-SMA, smad3, p-smad3, GAPDH primary antibodies were provided by CST. All values were normalized to GAPDH. All samples were analyzed in triplicate.
Histopathology
Mice liver samples were preserved in 4% paraformaldehyde solution to obtain histological slices. After fixation, all tissues were processed, paraplast-fixed blocks sectioned (5 mm thickness), and slides stained with hematoxylin and eosin (H&E), Masson and immunohistochemistry of α-SMA. Representative microphotographs were obtained using a light microscope (Nikon, Japan).
Collagen Determination
Collagen deposition in liver tissue Sects. (5–10 µm, paraffin embedded tissues) was localized by Masson’s trichrome staining using a commercially available staining kit according to the manufacturer’s instructions (Poly Scientific, Bay Shore, NY, United States). Fibrotic lesional density by area was measured on Masson’s trichrome stained sections and quantified by morphometric methodology.
Detection of ALT and AST
Serum levels of aspartate aminotransferase (AST) and aminotransferase (ALT) in mice were measured using ALT and AST kits (Nanjing Jiancheng Bioengineering Institute, Nanjing China) according to the manufacturer’s protocol.
Cells and Cell Culture
LX-2 human HSCs were obtained from Merck Millipore (Billerica, MA, USA) and were cultured in DMEM supplemented with 10% 10% fetal bovine serum (FBS) (Biological Industries, Kibbutz Beit-Haemek, Israel) at 37 °C with 5% CO2. Cells used in experiments were subjected to not more than 10 cell passages; 10 ng/ml TGF-β1 was used to induce target gene expression in LX-2 HSC. Exponentially growing cells were treated with TGF-β1 for 24 h. 0.9% saline was chosen as a vehicle to dissolve anlotinib. The anlotinib was added to the culture medium 2 h prior to addition of TGF-β1.
Cell Viability Determination
CCK-8 assay was used to measure LX-2 cell viability. Cells were plated at 5 × 103 cells per well in 96-well plates and exposed to different concentrations of anlotinib for 24 h. Then, the cells were incubated with 10 μL CCK-8 for another 2 h at 37 °C. The resulting product was measured at 450 nm using a microplate reader (Thermo Fisher Scientific).
Transwell Migration Assay
The migratory properties of LX-2 were assessed using a Transwell assay. Cells were seeded at a density of 4 × 104cells/well in the upper compartment of Transwell chambers with serum-free medium, and the lower compartment contained 700 μL of 10% Serum-containing medium per well. Migration was subsequently observed and measured.
Statistical Analysis
Data were analyzed using the Student’s t-test (Sigma Plot, SPSS Inc.) for differences between two groups, and are expressed as the mean ± SE. For comparisons among multiple groups, ANOVA (3-way) was performed, followed by t-test with Bonferronni correction using SAS 9.3 (SAS Institute Inc., Cary, NC, United States). All experiments were repeated at least 3 times. Differences were considered statistically significant at p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Results
To investigate the effects of anlotinib on hepatic fibrogenesis, a CCl4-induced hepatic fibrosis model was established in adult male C57BL/6 mice. After 4 weeks of either CCl4 or corn oil injection, the mice in the CCl4 group were randomly divided into two groups: one received CCl4 supplemented with normal saline and the other received CCl4 supplemented with anlotinib treated (oral, 3 mg/kg body weight, n = 5 in each group). After 2 weeks, the mice were sacrificed, and their livers were dissected and fixed. The degree of liver fibrosis was evaluated by quantifying Masson’s collagen areas in the liver tissue, The anlotinib-treated group showed significantly reduced liver inflammatory cell infiltration, fragmented hepatic nuclei, and collagen deposition compared with the CCl4 group (Fig. 1A, B). Additionally, anlotinib significantly attenuated the damage caused by CCl4. Notable reductions in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were observed in the anlotinib-treated mice (Fig. 1C, D). CCl4 cause F4 grade (Ishak) hepatic fibrosis, liver inflammatory scores ranged from 12 to 14 (Ishak), a mean ALT measurement of 130 U/L and a mean measurement AST value of 119 U/L in mice. However, the CCl4-induced changes were markedly attenuated by anlotinib treatment, which returned to F2 grade (Ishak) hepatic fibrosis, liver inflammatory scores ranged from 4 to 6 (Ishak), a mean ALT measurement of 40 U/L and a mean measurement AST value of 56 U/L in mice. These data indicated that anlotinib exerts a protective effect against CCl4-induced hepatic fibrosis and liver injury.

Anlotinib improved CCl4-induced liver fibrosis and liver function in mice. A The representative images of liver tissues (Oil, CCl4, CCl4 plus Anlotinib group) stained with H&E and Masson. Collagen deposition was indicated by the blue strands. The images were captured under a light microscope with 100 times and 200 times amplification. B Statistical analysis of Masson blue-positive collagen per field. C–D Serum ALT and AST levels in Oil, CCl4, CCl4 plus Anlotinib group. n = 5 in each group. *p < 0.05; ns, p > 0.05
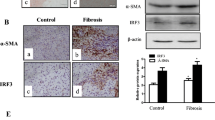
As shown in Fig. 2A, α-SMA, a widely accepted biomarker of HSC activation, and Col I, an important indicator of ECM deposition in the hepatic mesenchyme, were assessed. Immunohistochemistry showed α-SMA expression in the interstitial space of hepatocytes (Fig. 2A, B). In normal liver tissue (oil group), α-SMA was not expressed, while in fibrotic liver tissue (CCl4 group), it was highly expressed, as indicated by the large positive immunolocalization area in the liver. However, α-SMA expression was reduced when treated with anlotinib compared to that in the CCl4-only group (Fig. 2A, B). Furthermore, the protein levels of α-SMA, Col I, and p-Smad3 were significantly upregulated in the liver tissues of mice treated with CCl4 but returned to normal after anlotinib administration (Fig. 2C–G). These findings suggested that anlotinib ameliorated CCl4-induced liver fibrosis in mice by inhibiting the Smad3 pathway.

Anlotinib improved liver fibrosis via regulating TGF-β1/Smad3 pathway in CCl4-treated mice. A immunohistochemistry for α-SMA (brown area) in the liver tissues (Oil, CCl4, CCl4 plus Anlotinib group) under a light microscope with 200 times and 400 times amplification in mice. B Statistical analysis of α-SMA-positive ratio. C–G The representative western blot images of α-SMA, Collagen I (C) and p-smad3, smad3 (F) in liver tissues (Control, Anlitinib,CCl4,CCl4 plus Anlotinib group). The quantification of proteins α-SMA (D), Collagen I (E) by normalizing to the expression of GAPDH and proteins p-smad3 by normalizing to the expression of smad3 (G) with Image J. n = 5 in each group. ****p < 0.0001; **p < 0.01; *p < 0.05; ns, p > 0.05
We investigated the effects of anlotinib on TGF-β1-induced migration and proliferation of LX-2 cells in vitro. Initially, we determined the cytotoxicity of anlotinib in LX-2 cells using the CCK-8 assay, revealing cytotoxic effects at 2.5 μM (Fig. 3C). Therefore, we used a lower concentration of anlotinib (1 μM) in subsequent experiments. After 24 h of TGF-β1 treatment with or without anlotinib, we evaluated the migration of LX-2 cells in a transwell chamber. The results demonstrated that TGF-β1 treatment increased the migration of LX-2 cells; however, this effect was reversed by anlotinib administration (P < 0.0001; Fig. 3A, B). However, anlotinib alone did not affect the migratory ability of LX-2 cells in vitro.

Anlotinib inhibits migratory and proliferative ability of LX-2 in vitro. A The migratory ability of LX-2 (Control, Anlitinib, TGF-β1, TGF-β1 plus Anlotinib group) was measured under a light microscope at 200 times magnification (4 × 104 cells per well), LX-2 cells were treated with TGF-β1 (10 ng/ml) for 24 h or plus Anlotinib (1 μM). B Statistical analysis of migrating cell counts. C LX-2 cells were treated with indicated concentrations of Anlotinib for 24 h and cell viability was detected with CCK-8 kit. A density of 5 × 103 cells/well was planted in 96-well plate. n = 3 in each group. ****p < 0.0001; ***p < 0.001; *p < 0.05; ns, p > 0.05
We evaluated the effects of anlotinib on TGF-β1-induced liver fibrosis in vitro using LX-2 cells. As shown in Fig. 4A–C, anlotinib significantly reversed the expression of TGF-β1-induced Col I and α-SMA. However, treatment with anlotinib alone did not affect the expression of these proteins. These results suggest that anlotinib can improve TGF-β1-induced liver fibrosis in LX-2 cells. Subsequently, we investigated the effects of anlotinib on TGF-β1/Smad3 pathways in LX-2 cells. TGF-β1 treatment significantly increased p-Smad3 levels, but anlotinib treatment significantly attenuated this increase (Fig. 4D, E). These data demonstrate that anlotinib attenuated TGF-β1-induced in vitro liver fibrosis by inhibiting the TGF-β1/Smad3 pathway.

Anlotinib improved TGF-β1-induced liver fibrosis via regulating TGF-β1/Smad3 pathway in vitro. A–E The representative western blot images of α-SMA, Collagen I (A) and p-smad3, smad3 (D) in LX-2 cells treated with TGF-β1 (10 ng/ml) for 24 h or plus Anlotinib (Control, Anlitinib, TGF-β1, TGF-β plus Anlotinib group). The quantification of proteins α-SMA (B), Collagen I (C) by normalizing to the expression of GAPDH and proteins p-smad3 by normalizing to the expression of smad3 (E) with Image J. n = 3 in each group. ***p < 0.001; *p < 0.05; ns, p > 0.05
Discussion
Chronic liver injury, caused by various factors, is often accompanied by chronic liver inflammation. HSCs’ activation to become fibroblasts plays a crucial role in the pathogenesis of liver fibrosis, as they synthesize and deposit fibrous collagen, leading to liver fibrosis. Therefore, inhibition of HSC activation is a key therapeutic strategy for hepatic fibrosis [14, 17]. As liver fibrosis progresses, cirrhosis may occur, which may result in various complications of end-stage liver disease with a high fatality rate [7]. Therefore, an urgent need exists for drug research for the treatment of liver fibrosis. Although the cellular and molecular substrates of hepatic fibrosis are being increasingly studied and understood, many antifibrotic drugs and their mechanisms of action remain under investigation. In the past, some existing antitumor drugs, such as nintedanib, had drug targets similar to those of antifibrotic drugs and have been shown to be effective in improving fibrosis in in vivo and in vitro experiments [19]. Therefore, screening existing drugs for the treatment of other diseases to develop new antifibrotic drugs has certain advantages, including reduced research time and resources on drug safety and pharmacokinetics, which may help promote the clinical application of effective antifibrotic drugs [16].
Anlotinib is a small-molecule tyrosine kinase inhibitor used to treat various advanced tumors. CCl4 is a powerful hepatoxic and prooxidant agent which is widely used to induce acute liver injury, liver fibrosis, and cirrhosis models in recent years [33]. The damage-causing mechanism of CCl4 in tissues can be explained as oxidative damage caused by lipid peroxidation which starts after the conversion of CCl4 to free radicals of highly toxic trichloromethyl radicals (·CCl3) and trichloromethyl peroxyl radical (·CCl3O2) via cytochrome P450 enzyme [27]. In this study, we established a CCl4-induced mouse model of hepatic fibrosis based on the methods described by Domitrovic and Jakovac to evaluate the anti-fibrotic effect of anlotinib. Histopathological analysis of liver tissue sections from the CCl4-induced hepatic fibrosis model revealed that CCl4 caused hepatocyte necrosis and collagen matrix accumulation, resulting in the formation of interconnecting intervals and division of the liver parenchyma into independent fragments. This led to a significant increase in the area of hepatic fibrosis and the upregulation of α-SMA, a marker of HSC activation. However, treatment with anlotinib significantly normalized the structure of the hepatic lobules, reduced the infiltration of hepatic inflammatory cells, and reduced hepatic nucleus fragmentation, resulting in a reduced area of hepatic fibrosis and downregulation of α-SMA expression (Figs. 1 and 2A, B). We also measured the levels of ALT and AST, which are commonly used as liver injury enzyme markers [10]. These enzymes leak into the circulatory system after hepatocyte injury, indicating a change in membrane permeability and reflecting the integrity of the liver structure [18]. The AST and ALT levels in mice treated with CCl4 were significantly higher than those in the control group. However, treatment with anlotinib significantly reduced the increase in ALT and AST levels induced by CCl4, indicating its hepatoprotective activity (Fig. 1).
Western blot experiments on mouse liver proteins showed that the expressions of α-SMA, Col I, and p-Smad3 in the livers of the CCl4 group were significantly increased, whereas their expressions were significantly decreased by anlotinib. This suggests that anlotinib may reduce collagen synthesis by inhibiting the Smad3 signaling pathway in the liver (Fig. 2C–G).
TGF-β1 is currently the most effective pro-fibrotic cytokine and is considered a key factor in the development of liver fibrosis. TGF-β1 can induce the proliferation and activation of HSCs and promote myofibroblast formation and ECM deposition [28]. There is a large amount of preclinical evidence suggesting that some tyrosine kinase inhibitors, such as nintedanib and sorafenib, can strongly inhibit the TGF-β1/Smad3 signaling pathway [1, 13], suggesting a special relationship between the tyrosine kinase and TGF-β1 signaling pathways. Therefore, we examined whether the tyrosine kinase inhibitor anlotinib plays an antifibrotic role by blocking the TGF-β1/Smad3 signaling pathway. We established an in vitro cell model in which TGF-β1 induced LX-2 activation. The results showed that TGF-β1 significantly increased the expression of α-SMA and Col I in LX-2, indicating HSC activation. Anlotinib inhibited the increase of α-SMA and Col I induced by TGF-β1, suggesting that HSC activation might be prevented by anlotinib. We also demonstrated that anlotinib significantly inhibited TGF-β1-induced Smad3 phosphorylation (Fig. 4). Additionally, we conducted transwell assays to detect cell migration, and the results showed that TGF-β1 significantly increased the migration ability of LX-2, while anlotinib significantly inhibited the increase in TGF-β1-induced migration ability. Further studies are underway to clarify whether tyrosine kinases are involved in other mechanisms of fibrosis, which may lead to the development of additional therapeutic targets and drugs for liver fibrosis.
Conclusions
In conclusion, this study demonstrates, a regulation of TGF-β1 signaling by an FDA-approved drug—anlotinib—that prevents liver fibrosis and inflammation in a CCl4 induced liver fibrosis model, and the potential of anlotinib to suppress the profibrogenic activity of TGF-β1 signaling in vitro. Anlotinib may be useful for halting or reversing the progression of liver fibrosis and could be employed in the development of novel therapeutic drugs for the management of chronic liver diseases.
Data Availability
All raw data supporting our findings is available on request.
References
Adenina S, Louisa M, Soetikno V, Arozal W, Wanandi SI. The effect of alpha mangostin on epithelial-mesenchymal transition on human hepatocellular carcinoma HepG2 cells surviving Sorafenib via TGF-beta/Smad pathways. Adv Pharm Bull 2020;10:648–655.
Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol 2019;70:151–171.
Brenner DA. Reversibility of liver fibrosis. Gastroenterol Hepatol 2013;9:737–739.
Cai S, Wu L, Yuan S, Liu G, Wang Y, Fang L, Xu D. Carvacrol alleviates liver fibrosis by inhibiting TRPM7 and modulating the MAPK signaling pathway. Eur J Pharmacol 2021;898:173982.
Campana L, Iredale JP. Regression of liver fibrosis. Semin Liver Dis 2017;37:1–10.
Chen W, Zhang J, Zhong W, Liu Y, Lu Y, Zeng Z, Huang H, Wan X, Meng X, Zou F, Cai S, Dong H. Anlotinib inhibits PFKFB3-driven glycolysis in myofibroblasts to reverse pulmonary fibrosis. Front Pharmacol 2021;12:744826.
Friedman SL. Liver fibrosis—from bench to bedside. J Hepatol 2003;38:S38–S53.
He C, Wu T, Hao Y. Anlotinib induces hepatocellular carcinoma apoptosis and inhibits proliferation via Erk and Akt pathway. Biochem Bioph Res Co 2018;503:3093–3099.
Higashi T, Friedman SL, Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv Drug Deliver Rev 2017;121:27–42.
Lei F, Liu YM, Zhou F, Qin JJ, Zhang P, Zhu L, Zhang XJ, Cai J, Lin L, Ouyang S, Wang X, Yang C, Cheng X, Liu W, Li H, Xie J, Wu B, Luo H, Xiao F, Chen J, Tao L, Cheng G, She ZG, Zhou J, Wang H, Lin J, Luo P, Fu S, Zhou J, Ye P, Xiao B, Mao W, Liu L, Yan Y, Liu L, Chen G, Li H, Huang X, Zhang BH, Yuan Y. Longitudinal association between markers of liver injury and mortality in COVID-19 in China. Hepatology 2020;72:389–398.
Lei J, Li Q, Xu H, Luo M, Liu Z, Xiang D, Chen P. Anlotinib improves bile duct ligature-induced liver fibrosis in rats via antiangiogenesis regulated by VEGFR2/mTOR pathway. Drug Develop Res. 2022;84:143–155.
Liang L, Hui K, Hu C, Wen Y, Jiang X. Autophagy inhibition potentiates the anti-angiogenic property of multikinase inhibitor anlotinib through JAK2/STAT3/VEGFA signaling in non-small cell lung cancer cells. Ann Oncol 2019;30:ii4.
Liu F, Bayliss G, Zhuang S. Application of nintedanib and other potential anti-fibrotic agents in fibrotic diseases. Clin Sci 2019;133:1309–1320.
Lotersztajn S, Julien B, Teixeira-Clerc F, Grenard P, Mallat A. Hepatic fibrosis: molecular mechanisms and drug targets. Annu Rev Pharmacol 2005;45:605–628.
Martinez FJ, Collard HR, Pardo A, Raghu G, Richeldi L, Selman M, Swigris JJ, Taniguchi H, Wells AU. Idiopathic pulmonary fibrosis. Nat Rev Dis Primers 2017;3:17074.
Marti-Rodrigo A, Alegre F, Moragrega AB, Garcia-Garcia F, Marti-Rodrigo P, Fernandez-Iglesias A, Gracia-Sancho J, Apostolova N, Esplugues JV, Blas-Garcia A. Rilpivirine attenuates liver fibrosis through selective STAT1-mediated apoptosis in hepatic stellate cells. Gut 2020;69:920–932.
Pinzani M, Marra F. Cytokine receptors and signaling in hepatic stellate cells. Semin Liver Dis 2001;21:397–416.
Raja S, Ahamed KF, Kumar V, Mukherjee K, Bandyopadhyay A, Mukherjee PK. Antioxidant effect of Cytisus scoparius against carbon tetrachloride treated liver injury in rats. J Ethnopharmacol 2007;109:41–47.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, Cottin V, Flaherty KR, Hansell DM, Inoue Y, Kim DS, Kolb M, Nicholson AG, Noble PW, Selman M, Taniguchi H, Brun M, Le Maulf F, Girard M, Stowasser S, Schlenker-Herceg R, Disse B, Collard HR. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. New Engl J Med 2014;370:2071–2082.
Ruan H, Lv Z, Liu S, Zhang L, Huang K, Gao S, Gan W, Liu X, Zhang S, Helian K, Li X, Zhou H, Yang C. Anlotinib attenuated bleomycin-induced pulmonary fibrosis via the TGF-beta1 signalling pathway. J Pharm Pharmacol 2020;72:44–55.
Spagnolo P, Kropski JA, Jones MG, Lee JS, Rossi G, Karampitsakos T, Maher TM, Tzouvelekis A, Ryerson CJ. Idiopathic pulmonary fibrosis: disease mechanisms and drug development. Pharmacol Therapeut 2021;222:107798.
Sun M, Kisseleva T. Reversibility of liver fibrosis. Clin Res Hepatol Gas 2015;39:S60–S63.
Sun Y, Niu W, Du F, Du C, Li S, Wang J, Li L, Wang F, Hao Y, Li C, Chi Y. Safety, pharmacokinetics, and antitumor properties of anlotinib, an oral multi-target tyrosine kinase inhibitor, in patients with advanced refractory solid tumors. J Hematol Oncol 2016;9:105.
Syed YY. Anlotinib: first global approval. Drugs 2018;78:1057–1062.
Taurin S, Yang CH, Reyes M, Cho S, Coombs DM, Jarboe EA, Werner TL, Peterson CM, Janat-Amsbury MM. Endometrial cancers harboring mutated fibroblast growth factor receptor 2 protein are successfully treated with a new small tyrosine kinase inhibitor in an orthotopic mouse model. Int J Gynecol Cancer 2018;28:152–160.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastro Hepat 2017;14:397–411.
Unsal V, Cicek M, Sabancilar I. Toxicity of carbon tetrachloride, free radicals and role of antioxidants. Rev Environ Health 2021;36:279–295.
Vallee A, Lecarpentier Y, Vallee JN. Thermodynamic aspects and reprogramming cellular energy metabolism during the fibrosis process. Int J Mol Sci 2017;18:2537.
Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B. Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 2014;349:209–220.
Xia X, Pi W, Lan Y, Wu X, Lv D, Meng Y, Yang H, Wang W. Synergistic antitumor effects of anlotinib combined with oral 5-fluorouracil/S-1 via inhibiting Src/AKT signaling pathway in small-cell lung cancer. Anal Cell Pathol 2022;2022:4484211.
Xie C, Wan X, Quan H, Zheng M, Fu L, Li Y, Lou L. Preclinical characterization of anlotinib, a highly potent and selective vascular endothelial growth factor receptor-2 inhibitor. Cancer Sci 2018;109:1207–1219.
Yang YR, Bu FT, Yang Y, Li H, Huang C, Meng XM, Zhang L, Lv XW, Li J. LEFTY2 alleviates hepatic stellate cell activation and liver fibrosis by regulating the TGF-beta1/Smad3 pathway. Mol Immunol 2020;126:31–39.
Yao Q, Lin Y, Li X, Shen X, Wang J, Tu C. Curcumin ameliorates intrahepatic angiogenesis and capillarization of the sinusoids in carbon tetrachloride-induced rat liver fibrosis. Toxicol Lett 2013;222:72–82.
Yuan M, Guo XL, Chen JH, He Y, Liu ZQ, Zhang HP, Ren J, Xu Q. Anlotinib suppresses proliferation, migration, and immune escape of gastric cancer cells by activating the cGAS-STING/IFN-beta pathway. Neoplasma 2022;69:807–819.
Zhan L, Huang C, Meng XM, Song Y, Wu XQ, Yang Y, Li J. Hypoxia-inducible factor-1alpha in hepatic fibrosis: a promising therapeutic target. Biochimie 2015;108:1–7.
Zhao XK, Yu L, Cheng ML, Che P, Lu YY, Zhang Q, Mu M, Li H, Zhu LL, Zhu JJ, Hu M, Li P, Liang YD, Luo XH, Cheng YJ, Xu ZX, Ding Q. Focal adhesion kinase regulates hepatic stellate cell activation and liver fibrosis. Sci Rep-UK 2017;7:4032.
Zhou WC, Zhang QB, Qiao L. Pathogenesis of liver cirrhosis. World J Gastroentero 2014;20:7312–7324.
Acknowledgments
This work was supported by the Science and Technology Fund Project of Guizhou Provincial Health Commission (contract numbers gzwkj2021-074 to Ye-Ting Wu) , the National Natural Science Foundation of China (grant numbers 82060116 and 82260129 to Xue-Ke Zhao) and the Science and Technology Support Project of Guizhou Province, No. [2021] 094. We would like to thank Editage (www.editage.cn) for English language editing.
Funding
This research was funded by Science and Technology Fund Project of Guizhou Provincial Health Commission (contract numbers gzwkj2021-074 to Ye-Ting Wu), the National Natural Science Foundation of China (grant numbers 82060116 and 82260129 to Xue-Ke Zhao) and the Science and Technology Support Project of Guizhou Province(No. [2021] 094).
Author information
Authors and Affiliations
Contributions
Conceptualization: XKZ, WFZ. Data curation: YTW, QZL, XKZ, MM, GLZ, WFZ. Formal analysis: YTW, QZL. Methodology and Resources: YTW, XKZ, WFZ. Project administration and Writing – original draft: YTW. Software: QZL. Supervision: WFZ. Visualization: YTW, WFZ. Writing – review & editing: YTW, QZL, XKZ.
Corresponding author
Ethics declarations
Conflict of interest
The authors report no conflicts of interest in this work.
Ethics approval
All experiments involving animals should obtain relevant ethics approval in accordance with national and institutional guidelines prior to commencing any animal work. This study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the ethics committee of Guizhou Medical University, Guizhou, China. The animal experimental ethical number is 2000732.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, YT., Li, QZ., Zhao, XK. et al. Anlotinib Attenuates Liver Fibrosis by Regulating the Transforming Growth Factor β1/Smad3 Signaling Pathway. Dig Dis Sci 68, 4186–4195 (2023). https://doi.org/10.1007/s10620-023-08101-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-023-08101-1