
Abstract
Inflammatory bowel disease (IBD) is a chronic and nonspecific intestinal inflammatory condition with high relapse rate. Its pathogenesis has been linked to dysbacteriosis, genetic and environmental factors. In recent years, a new type of lymphocytes, termed innate lymphoid cells, has been described and classified into three subtypes of innate lymphoid cells—group 1, group 2 and group 3. An imbalance among these subsets’ interaction with gut microbiome, and other immune cells affects intestinal mucosal homeostasis. Understanding the role of innate lymphoid cells may provide ideas for developing novel and targeted approaches for treatment of IBD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD) is a complex disorder comprising two main phenotypes, Crohn’s disease (CD) and ulcerative colitis (UC). IBD is characterized by high chronic relapse rates, with acute episodes and intervals of remission [1,2,3,4]. Its global incidence has significantly increased in recent years. IBD patients experience complications such as fistula, abdominal abscess and intestinal obstruction due to poor drug efficacy. In addition, prognosis and quality of life of IBD patients are extremely poor which calls for development of new therapeutic strategies and targets to combat the disease [5]. IBD is induced by environmental, genomic, microbial and immunological factors [6]. Studies on pathogenesis of this disease have focused on CD4-positive (CD4+) T lymphocytes, including their different subgroups such as type 1T helper (Th1), type 17 (Th17) and type 2 (Th2) cells. Th1, which expresses transcription factor T-bet and cytokines IFN-γ, and Th17, which expresses the transcription factor RORγt and cytokines IL-17, have been found to be enriched in the inflamed mucosa of IBD patients [7,8,9], while Th2 expressing transcription factor GATA-3 and cytokines IL-5 and IL-13 have also been implicated in the development of IBD [10, 11]. Recent reports have demonstrated the role played by innate lymphoid cells (ILCs), a novel type of innate lymphocytes, in intestinal mucosal homeostasis and development of IBD [12,13,14]. The ILC family plays an important role in body’s immune system, which not only provide defense against pathogen invasion and infection, but also contributes to organ formation, tissue repair and mucosal homeostasis [15, 16]. For a long time, natural killer (NK) cells have been the only members of the innate immune system known to belong to the lymphoid lineage. However, since the discovery of lymphoid tissue-inducing cells (LTi cells) in 1997, the population of lymphocytes in immune system has expanded [17]. ILCs have a similar functional diversity to CD4+ T cells [18,19,20,21]. The ILC family is further divided into three subgroups, group 1 ILCs (compromising NK and ILC1s), group 2 ILCs (ILC2s) and group 3 ILCs (ILC3s), depending on secretion level of cytokines and expression of transcription factors [22,23,24]. This review describes the role of ILCs in IBD. Particularly, we focus on the function of ILCs in regulating homeostasis and discuss potential therapeutic strategies for targeting ILCs in IBD.
Origin and Classification of ILC
Innate lymphoid cells are derived from common lymphoid progenitors (CLPs), which mainly express transcription factors such as IL-2R, Thy1 (CD90), IL-2Rα (CD25), IL-7Rα (CD127), Sca1 and c-kit (CD117) [22]. CLPs differentiate into T, B cell precursors [25], and this is dependent on E2A, E2-2, HEB and EBF-1. They then differentiate into co-ILC precursor cells (CILP) under the influence of Id2, TCF1, NFIL3 and Tox. CILPs develop into NK precursor cells (NKP) and common helper ILC progenitor cells (CHILP) under the influence of CXCR6 and α4β7, while CHILP develops into 3 ILC variants (group 1, group 2 and group 3 ILCs), whereas NKP gives rise to classical NK cells.
Group 1 ILCs
ILC1s and NK cells in human and mouse express T-bet, while Eomes are expressed only in mouse NK cells. NK cells are similar to CD8+ T cells while ILC1s are closely related to Th1 cells [26]. ILC1s express CD127, and both NK and ILC1s express CD122 (β chain of IL15 and IL2 receptor), as well as NK cell receptors NK1.1 and NKp46. In terms of distribution, NK cells are mainly found in the blood, cord blood, bone marrow, spleen and the lungs, whereas ILC1s mainly reside in colon tissues and the tonsil [13, 27]. The human gut has two major subpopulations of ILC1s: (i) the lamina propria ILC1s, which express CD161 and CD127, but lack NKp44 [28], and (ii) intraepithelial ILC1s that express NKp44 and CD103 but lack CD127, and are similar to classical NK cells in phenotype and cytotoxicity [29]. NK cells and ILC1s respond to IL15, IL12 as well as IL18 and express IFN-γ and TNF when activated. For this reason, they protect against intracellular microbes, viruses and tumors [25, 30]. In addition, NK cells secret granzyme B and perforin, which promote apoptosis of infected cells [31, 32] (Table 1).
Group 2 ILCs
Transcription factor GATA3 is required for the maintenance and normal functioning of mature group 2 ILCs (ILC2s) in humans and mice [33, 34]. In addition, other transcription factors such as, TCF1, Bcl-11b, ETS1 and Id2 have been found to play important roles in differentiation and prototypical stability of mouse ILC2s [35, 36]. In terms of distribution in humans and mice, ILC2s are abundant in fetal gut; however, they only account for 0.01–0.1% of intestinal hematopoietic cells in adults. Moreover, ILC2s are abundant in fat, spleen, mesenteric lymph nodes, lungs and skin, and tonsil. They are similar to Th2 cells, which secrete IL-4, IL-5, IL-6, IL-9, IL-13 and GM-CSF under the activation of IL-2, IL-25, IL-33, PGD2, TL1A and TSLP [37, 38]. In addition, ILC2s promote airway inflammation, maintain and repair airway epithelium, induce atopic dermatitis and protect the body against helminth infection [39,40,41] (Table 1).
Group 3 ILCs
Continuously expressing RORγt and aryl hydrocarbon receptor (AHR) are essential for the survival and differentiation of ILC3s [42, 43]. These cells are the main ILC subsets in intestinal tissues. Group 3 ILCs (ILC3s) are divided into two subgroups, CCR6+ ILC3s and CCR6− ILC3s, based on expression of chemokine receptor 6 (CCR6) [23]. CCR6+ ILC3s subpopulation, including embryonic and mature LTi cells, express IL-22, IL-17, IgA and lymphotoxin [23, 44,45,46]. Based on expression of natural cytotoxicity triggering receptor (NCR), NKp44 (NCR2) in humans and NKp46 (NCR1) in mice, CCR6− ILC3s are further divided into NCR+ ILC3s and NCR− ILC3s. LTi cells are mainly distributed in the intestinal and lymphoid tissues, whereas NCR+ ILC3s and NCR− ILC3s are abundant in the skin and intestinal lamina propria [47, 48]. Moreover, NCR+ ILC3s resemble Th22 cells and are capable of producing IL-22 in response to cytokines like IL-1β, IL-23 and IL-2. On the other hand, NCR− ILC3s are similar to Th17 cells and express cytokines IL-17 and GM-CSF in response to cytokines such as IL-6 [49,50,51,52]. Functionally, ILC3s resist gastrointestinal inflammation, maintain intestinal mucosal homeostasis, and contribute to the pathological response against IBD [45, 53,54,55,56,57]. (Table 1).
Balance Between ILC Subsets and IBD
Relationship Between ILC1/ILC3 Balance and IBD
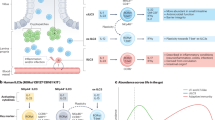
An alteration between ILC1s and ILC3s occurs in IBD. IL-12 drives the transdifferentiation of ILC3s into ILC1s and produce IFN-γ, which participates in inflammatory responses in the intestinal mucosa [58]. However, this transdifferentiation is reversible, whereby ILC1s transdifferentiate into ILC3s when stimulated by IL-2, IL-23 and IL-1β. Dendritic cells (DCs) regulate the plasticity of ILC3s and ILC1s. Studies have demonstrated that CD14− DCs promote expression of c-kit and NKp44 in ILC1s and cause them to transdifferentiate into NCR+ ILC3s in vitro, whereas CD14+ DCs promote the conversion of NCR+ ILC3s to ILC1s [59]. Furthermore, a mutual transformation between two ILC3 subsets (NCR+ ILC3s and NCR− ILC3s) has been reported. A study by Klose et al. [47] found that NCR− ILC3s (NKp46−CCR6−RORγt+ ILCs) upregulate T-bet expression and transdifferentiate into NCR+ ILC3s (NKp46+CCR6−RORγt+ ILCs) under the influence of interleukin IL-23 in a mouse model of Typhoid bacillus infection. Bernink et al. [28] reported that NCR− ILC3s (Lin−CD127+c-Kit−NKp44−ILCs) isolated from human tonsils and fetal gut differentiated into NCR+ ILC3s (NKp44+ ILC3s) under IL-23 and IL-1β stimulation in vitro. Similar results were reported by Teunissen et al. [60] in psoriasis patients, in which NCR− ILC3s isolated from the skin transformed to NCR+ ILC3s under exogenous stimulation by IL-1β and IL-23 cytokines. This conversion may be the cause of elevated expression of IL-22 and accumulation of NCR+ ILC3s in the skin of psoriasis patients. High TGF-β expression reverses this transformation by promoting the conversion of NCR+ ILC3s to NCR− ILC3s. In vitro experiments have revealed that NCR− ILC3s are converted to NCR+ ILC3s under the action of Notch in the absence of TGF-β expression [61, 62]. The dynamic balance between NCR+ ILC3s and NCR− ILC3s maintains intestinal antibacterial immunity and intestinal mucosal integrity, while an imbalance between the two subsets promotes development of intestinal inflammation [63] (Fig. 1).

Innate lymphoid cells (ILCs) in the gut and the plasticity of ILCs. The increase of ILC1s is accompanied by a large decrease of NCR+ ILC3s and increase of NCR− ILC3s in the inflamed intestine tissues of Crohn’s disease (CD) patients. The plasticity of ILC1s/ILC3s: interleukin (IL)-12 or CD14+ dendritic cells (DCs) drives ILC3s to transdifferentiate into interferon (IFN)-γ-producing ILC1s participating inflammatory response in inflamed intestinal mucosa. The plasticity of ILC1s to ILC3s is reversible in response to IL-2, IL-23 and IL-1β, besides, CD14− DCs upregulate c-kit and NKp44 expression in ILC1s and then differentiate into ILC3s. Furthermore, a mass of transforming growth factor (TGF)-β promotes the transformation of NCR+ ILC3s to NCR− ILC3s, while Salmonella typhi in a mouse model of Typhoid bacillus infection upregulate T-bet expression in NCR− ILC3s and then transdifferentiate into NCR+ ILC3s. The plasticity of ILC2s: IL-12 results in the conversion of ILC2s into IFN-γ-producing ILC2s, which is high in the inflamed gut of CD patients. And part of ILC2s appear to loss IL-13 expression adopt the feature of ILC1s in the presence of Il-12. AHR activation suppresses ILC2s function but enhances ILC3s function
Relationship Between ILC2/ILC1, ILC2/ILC3 and IBD
The intestinal mucosa of CD patients contains a high proportion of IL-13-producing ILC2s, which produce IFN-γ. Treatment with IL-12 in vitro decreases IL-13 expression in ILC2 which then resembles ILC1 (IL-13−IFN-γ+) [64]. Besides, recent studies have shown that deletion of AHR enhances the function of gut ILC2s and contributes to anti-helminth immunity, whereas its activation suppresses ILC2s function but enhances ILC3s function, thereby contributing to antibacterial immunity [65]. It is possible that ILC2/ILC3 transformation mediated by changes in intestinal adopted AHR expression affects intestinal immune response in IBD (Fig. 1).
A balance in ILCs levels is, therefore, important for intestinal homeostasis, with abnormal levels potentially contributing to colitis in patients with IBD.
ILC Regulates Intestinal Homeostasis
Different ILC subsets maintain a balance and contribute to intestinal homeostasis under physiological conditions. Abnormal proportion of ILC subsets disrupts intestinal homeostasis leading to inflammation in the gut. Indeed, intestinal tissues of CD patients have high levels of ILC1 accompanied by a decrease in NCR+ ILC3s, which increase the severity of the disease [28, 29, 66]. Patients with IBD have high numbers of NCR− ILC3s, IL17 and IFN-γ. In addition, the patients exhibit high neutrophils recruitment, and activation of macrophages, all of which exacerbate intestinal inflammation. Meanwhile, the number of NCR+ ILC3s and levels of IL22, RegIIIγ, RegIIIb, Fut2 and mucin protein Muc2 are all decreased in patients with IBD [67, 68]. These patients have low expression of the tight junction protein, claudin-2 and decreased regeneration of intestinal epithelial cells [51, 69], implying that mucosal barrier integrity and homeostasis in the intestinal tract are damaged.
The role of ILC2s in immune imbalance in IBD remains undefined. Kobori et al. [70] found that the IL33-ILC2-AREG-EGFR pathway was dysregulated in the intestinal mucosa of IBD patients. This weakened intestinal epithelial defense system, which in turn caused intestinal inflammation. Administration of exogenous IL-33 or ILC2s may decrease the degree of mouse intestinal inflammation [71, 72]. However, other studies have indicated that IL-33 is highly expressed in intestinal mucosa of patients with IBD where it promotes colitis [70, 73], which is contrary to the above results. Besides, in UC patients, levels of ILC2s are increased in inflammatory tissues of intestinal mucosal compared with non-inflammatory sites and non-IBD controls [74]. IL-13-producing ILC2s are elevated in oxazolone-induced mouse UC model [75], indicating that ILC2s might play a proinflammatory role in UC. Further studies are needed to resolve these contradictions.
Studies have shown that AHR expression, which regulates the production and function of ILC3s in intestinal immunity, is downregulated in patients with IBD [76]. In fact, deletion of AHR in Ahr−/− mice resulted in an increase in intestinal Th17 cells, suggesting that ILC3s may negatively regulate Th17 cells. Elsewhere, downregulation of IL-22 in Ahr−/− mice resulted in proliferation of symbiotic split filamentous bacteria (SFB), which promoted the proliferation of Th17 cells. AHR ameliorates T cell-mediated experimental colitis by inhibiting pathogenic Th17 cells [77]. A complex balance between ILCs and Th17 cells, regulated by AHR and commensal flora, is also essential for intestinal homeostasis.
Interaction of the Gut Microbes and ILCs
Microbiota regulate the interactions between ILCs and the host [78,79,80]. Most studies on ILCs involved with microbes have focused on ILC3s.
Signals generated by microbiota play an essential role in maturation and functioning of ILC3s [81]. Notably, studies have shown that production of IL-22 by ILC3s is decreased in germ-free or antibiotic-treated mice [82]. Meanwhile, microbiota symbiosis is disrupted when IL22 is depleted or decreased [83, 84]. Commensal bacteria increase the secretion of IL22 from ILC3, which promotes host resistance/defense to pathogens [68, 85]. High-fat diet has been found to alter intestinal microbiota (marked by amplification of Firmicutes) and lead to increased proinflammatory IL-17-producing NCR− ILC3s and reversed by IL-17 blockade [86]. Apart from IL-22 and IL-17, ILC3s is also a major source of IL-2. In fact, IL-2 produced by ILC3s is significantly reduced in the small intestine of CD patients and is associated with low frequencies in the number of Treg cells. Studies have shown that microbiota-dependent axis and IL-1β-dependent axis promote IL-2 production by ILC3s and coordinate immune function in the gut [87], suggesting a unique role for ILC3s in the interaction between microbiota and intestinal immunity. Additionally, ILC3s secrete lymphotoxin α (TNFβ) which is required for IgA production and intestinal commensal homeostasis [45, 80]. Microbiota trigger the activation and secretion of ILC3s and maintain the intestinal homeostasis in IBD.
Microbiota regulate body composition, including ILC3s, through a biological clock transcription molecule. Studies have reported that expression of transcription factor, Nfil3, in intestinal epithelial cells changes daily and that the amplitude of the circadian clock is regulated by microorganisms through ILC3s, STAT3, epithelial cell circadian clock as well as host metabolism [88]. Recently, intestinal ILC3 and its associated homeostasis in mice were found to be regulated by light-entrained and brain-tuned circadian circuits. Ablation of ILC3 circadian regulator was further implicated in disruption of gut homeostasis. Furthermore, dysregulation of brain rhythmicity brought about a disruption in circadian ILC3 vibrations, a deregulated microbiome, disrupted lipid metabolism and impaired intestinal homeostasis [89]. Intestinal ILC3s are enriched during expression of circadian-related genes. For instance, BMAL1-deficient ILC3s showed increased proapoptotic pathways, while depletion of microbiota by antibiotics was found to restore cellular homeostasis in the gut. ILC3s from inflamed mucosa of IBD exhibited variations in expression of several circadian-related genes [90]. Therefore, gut commensal microbiota alters the activation of ILC3s by regulating circadian rhythm during the pathogenesis of IBD.
Microbiota may also influence activation of ILC2s in intestinal immunity. IL-25, produced in a microbial-dependent manner, activates ILC2 to enhance intestinal barrier [82, 91]. IL-33 promotes activation of ILC2 and prevents Clostridium difficile colitis [92]. In addition, specific deletion of T-bet in ILCs promotes the development of ILC2s and protects the host from Trichinella spiralis infection and inflammatory colitis [93]. How microbiota regulate ILC1 or ILC2 in the mucosal immunology of IBD is not clear.
Microbiota-associated components and their associated metabolites also play a role in the functioning of ILCs. Kinnebrew et al.[67] found that IL23 secreted by flagellin-stimulated dendritic cells promote IL22 expression in ILC3s and that IL22 further acts on IECs to secrete antibacterial peptides that enhance long-term tolerance to ingested antigens. Recent metabolomic studies targeting microbiota in IBD have focused on short-chain fatty acids (SCFAs), bile acid and tryptophan metabolism. Diet-derived SCFAs metabolized by symbiotic microbiota promote the proliferation of ILCs in the intestines by regulating G protein-coupled receptors (GPCRs) on ILCs, including ILC1, ILC2 and ILC3 [94]. How microbial-related bile acid metabolism regulates ILC in IBD is unclear. Dysbiosis of the intestinal flora in IBD impairs tryptophan metabolism by affecting several pathways, e.g., by upregulating the kynurenine pathway and inhibiting the indole pathway [95, 96]. This downregulates AhR, impairs intestinal barrier integrity and decreases secretion of IL-22 to promote intestinal inflammation [96, 97]. It is reported that ILC3s are a dominant source of IL-22 in the steady state in the presence of commensal bacteria [83].Therefore, we speculate that the disrupted indole pathway in tryptophan metabolism may downregulate the function of ILC3 and accelerates intestinal inflammation in IBD, but this finding needs to be verified in further studies.
Other Intestinal Immune Cells Interacting with ILCs in IBD
ILCs lack adaptive antigen receptors. On the other hand, myeloid cell lineages (DCs, mononuclear phagocytes) and epithelial cells are able to sense microbial or viral infections and/or tissue damage and produce cytokines that interact with ILCs [98].
The secretion of cytokines IL-1β and IL-23 by local DCs, enhanced by serum amyloid A from intestinal epithelial cells, regulates the production of IL-22 by intestinal NCR-ILC3 [99, 100]. Besides IL-23, IL-1β expressed mainly by DCs, other mediators related to the regulation of ILC3s have been identified. For instance, some studies have shown that RA, a metabolite of vitamin A, relieves intestinal inflammation of DSS-induced colitis or Citrobacter rodentium infection. RA treatment promotes production of IL-22 by ILC3s and γδT cells, which plays a role in production of antibacterial peptides, RegIIIβ and RegIIIγ, in the colon and promotes repair of the intestinal mucosa [101]. IL-1β, IL-23 or RA produced by DCs in IBD are important stimulators that activate ILC3s.
The CX3CR1+ mononuclear phagocytes (MNPs) in mouse and human tissues are more effective, than DCs, in promoting ILC3-mediated production of IL-22 in vitro and in vivo [102]. It is hypothesized that CX3CR1+ MNPs play a role in integrating immune signals during regulation of colonic ILC3s in IBD. One of the products of MNPs, TL1A, promotes ILC3-centered barrier immunity and prevents acute colitis. In addition, TL1A-dependent secretion of OX40L expression of ILC3s stimulates T cell activation during chronic colitis [103]. TL1A has also been found to increase ILC3s’ ability to secrete GM-CSF to support mucosal protection [102]. Interestingly, GM-CSF promotes secretion of IL-10, RA of MNPs, and induces differentiation of Treg cells to maintain intestinal immune tolerance and intestinal homeostasis [104], although studies have also implicated IL-23R/GM-CSF axis within ILC3s in promoting of recruitment of neutrophils and causing increased intestinal inflammation [52]. Here, we reveal that the production of GM-CSF by ILC3s enhanced by MNPs can be used as a double-edged sword, which is depend on the different context and different upstream activators.
Apart from MNPs and DCs, epithelial cells that interact with ILCs and regulate intestinal homeostasis have been identified. For instance, Levy et al. [105] found that under steady-state conditions, IL-18 secretion of epithelial cells is enhanced by some metabolites including taurine-bound bile acids, carbohydrates and long-chain fatty acids, and these can activate the NLRP6 inflammasome, thereby increasing expression of IL-22 from ILC3s to regulate host–microbial interactions and maintain intestinal homeostasis. However, IL-18 from epithelial cells also showed proinflammatory effect. In a Toxoplasma gondii infection mouse model, ILC3-derived IL-22 in inflamed mucosa was found to induce production of inflammasome-dependent IL-18. Increased IL-18 was reported to amplify uncontrolled production of IL-17 and IL-22 and further cause damage to the intestinal mucosa [106]. Overall, the epithelial inflammasome is one of the key regulators in the intestine; inflammasome activation and IL-18 secretion mediate intestinal homeostasis or inflammation through ILC3s.
In addition, ILCs could be directly regulating intestinal immune cells, according to some studies. Particularly, ILC3-intrinsic expression of major histocompatibility complex class II (MHCII) induces the death of activated commensal bacterial specific T cells (CD4+ T cells) and maintains intestinal immune tolerance in commensal bacteria [107, 108].
Collectively, the interaction between ILC and other intestinal immune cells determines the activation and function of ILC directly and regulates intestinal mucosal immunology in IBD.
Clinical Importance of ILCs
Anti-inflammatory, immunomodulatory and immunosuppressive drugs as well as biologic agents like TNF blockers (infliximab) and vedolizumab have been applied to manage IBD [109]. Different kinds of therapeutic strategies have been practiced for clinical treatment of IBD (Fig. 2), but biological therapies are required to be studied further.

Therapeutic targets associated with ILCs in IBD. A number of current and potential future targets provide therapeutic effect in IBD. These targets involves in the generation, activation or effective function of ILCs and T cells. (*, FDA-approved and providing clinical benefit; #, efficacy exhibited in clinical trials; †, poor efficacy in clinical trials; ‡, promising preclinical target in mouse models.)
Th17 cells and ILC3s have numerous similar expression profiles of transcription factors including those related to RORγt and similar cytokines like IL-17. These factors exert different functions in immune response, inflammation and tissue damage repair. For example, when a Citrobacter rodentium-infected mouse model was treated with GSK805, a biological factor that inhibits expression of transcription factor RORγt, the number of Th17 cells was significantly downregulated compared with those in control group. Consequently, intestinal inflammation was alleviated. Studies have shown that transient inhibition of RORγt expression, using biological agents, may provide a potential strategy for treatment of intestinal inflammation [110]. However, secukinumab which is IL-17A monoclonal antibody resulted in undesirable outcomes in the clinical multicenter phase 2a study [111] and is therefore considered ineffective in CD patients owing to exacerbation of the disease. This is attributed to the effect of the compound on protective function of IL-17 as well as increased Th1 and IFN-γ-mediated inflammatory responses [111,112,113,114]. The findings of this study indicate the complexity of the IL23/IL17 axis, in which IL-17 may be one of the IL23-induced redundant mediators. Ustekinumab, a monoclonal antibody therapy against the p40 subunit of IL23, has been approved by the U.S. Food and Drug Administration and is in use for the treatment of moderate to severe CD [115]. In addition, a phase 2 randomized trial on patients with UC found that mirikizumab (a monoclonal antibody against the p19 subunit of IL23) could induce a clinical remission after 12 weeks, although the optimum dose remains to be determined [116]. This clinical trial indicated that IBD is not primarily caused by IL-17A-mediated intestinal inflammation. Besides, IL-17A and IL-17F have different functions. For instance, IL-17A promotes host immunity to fungal and bacterial pathogens. A dysregulated IL-17A can aggravate inflammation and promote development of inflammatory diseases, such as inflammatory bowel disease [117]. On the other hand, IL-17F acts as a pro-inflammatory factor in colitis, promoting the production of antimicrobial peptides and IL-22. A recent study showed that antibodies to IL-17F, but not IL-17A, ameliorated DSS-induced colitis [118], suggesting that IL-17F is a potential target for IBD treatment.
In addition, IFN-γ is a major cytokine effector in ILC1s and can further be co-expressed by some ILC3s subpopulations. Targeted neutralization of IFN-γ, fontolizumab (a monoclonal antibody against IFN-γ), has been evaluated in some CD patients, as described in a clinical phase 2 trial [119]. Although this study did not meet the expected criteria (remission at week 4), the results therein showed significant alleviation of clinical symptoms and increased rates of the associated remission after the study period.
Generally, ILC1s and ILC3s as well as other intestinal immune cells including mononuclear phagocytic cells secrete TNF [120]. An anti-TNF therapy has been successfully applied for treatment of IBD [121]. The authors observed downregulation of IL-22BP, and upregulation of IL-22, in CD4+ T cells after treatment of IBD patients with TNF-α antibodies [122]. This could be one of the mechanisms for TNF-α antibody treatment.
Moreover, cytokine IL-6, together with IL-23 and IL-1, promotes the expression of IL-17A of ILC3s. IL-6 antibodies alleviate enteritis in TRUC mice and inhibit IL-17A production. A recent randomized trial reported that anti-IL-6 antibody (PF-04236921) could induce clinical remission in moderate-to-severe CD patients following anti-TNF treatment failure [123]. This indicates that IL-6 can be used to develop IBD treatments.
Another approach for treating ILC-mediated IBD entails reducing the number of intestinal ILC, through consumption of intestinal ILC or prevention of its location in the intestine. Methods that consume ILC, such as monoclonal antibodies against CD90, have resulted in remarkable efficacy in IBD animal models [124]. However, related clinical trials are yet to be conducted; hence, there is still much to do to identify novel and effective targets for clinical use.
Conclusions and Perspectives
Innate lymphoid cells regulate intestinal homeostasis and pathological processes of IBD. Understanding the imbalance in ILC subgroups, and how they regulate intestinal homeostasis is key to developing novel approaches for the treatment of IBD. Some treatment therapies that incorporate molecules or cells involved in the pathogenesis of the disease, including blockers of IL-6 and IFN-γ, as well as activators of IL-33, IL-22 and MHCII+ ILCs expression, have shown promising efficacies in animal experiments. However, further clinical trials are needed to determine their clinical value. Further research on ILCs should be conducted to increase our understanding of the pathogenesis of IBD and generate novel strategies for its treatment.
References
Liu TC, Stappenbeck TS. Genetics and pathogenesis of inflammatory bowel disease. Annu Rev Pathol. 2016;11:127–148
Peloquin JM, Goel G, Villablanca EJ, Xavier RJ. Mechanisms of pediatric inflammatory bowel disease. Annu Rev Immunol. 2016;34:31–64
Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel J-F. Ulcerative colitis. Lancet. 2017;389:1756–1770
Torres J, Mehandru S, Colombel JF, Peyrin-Biroulet L. Crohn’s disease. Lancet (London, England). 2017;389:1741–1755
Uhlig HH, Powrie F. Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu Rev Immunol. 2018;36:755–781
de Souza HSP, Fiocchi C, Iliopoulos D. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. 2017;14:739–749
Breese EB, Corrigan CP, Walker-Smith CJ, MacDonald JA. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology. 1993;78:127–131
Rovedatti L, Kudo T, Biancheri P et al. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut. 2009;58:1629–1636
Liu H, Dasgupta S, Fu Y et al. Subsets of mononuclear phagocytes are enriched in the inflamed colons of patients with IBD. BMC Immunol. 2019;20:42
Park JH, Jeong DY, Peyrin-Biroulet L, Eisenhut M, Shin JI. Insight into the role of TSLP in inflammatory bowel diseases. Autoimmun Rev. 2017;16:55–63
Giuffrida P, Caprioli F, Facciotti F, Di Sabatino A. The role of interleukin-13 in chronic inflammatory intestinal disorders. Autoimmun Rev. 2019;18:549–555
Ealey KN, Koyasu S. How many subsets of innate lymphoid cells do we need? Immunity. 2017;46:10–13
Simoni Y, Newell EW. Dissecting human ILC heterogeneity: more than just three subsets. Immunology. 2018;153:297–303
Giuffrida P, Corazza GR, Di Sabatino A. Old and new lymphocyte players in inflammatory bowel disease. Dig Dis Sci. 2018;63:277–288. https://doi.org/10.1007/s10620-017-4892-4
Cupedo T, Crellin NK, Papazian N et al. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10:66–74
Cella M, Fuchs A, Vermi W et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–725
Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity. 1997;7:493–504
Huang Y, Mao K, Germain RN. Thinking differently about ILCs-Not just tissue resident and not just the same as CD4(+) T-cell effectors. Immunol Rev. 2018;286:160–171
Trabanelli S, Gomez-Cadena A, Salomé B et al. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping. Cytometry B Clin. Cytometry. 2018;94:392–399
Panda SK, Colonna M. Innate lymphoid cells in mucosal immunity. Front Immunol. 2019;10:861
Sonnenberg GF, Hepworth MR. Functional interactions between innate lymphoid cells and adaptive immunity. Nat Rev Immunol. 2019;19:599–613
Spits H, Artis D, Colonna M et al. Innate lymphoid cells: a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149
Zook EC, Kee BL. Development of innate lymphoid cells. Nat Immunol. 2016;17:775–782
Lim AI, Verrier T, Vosshenrich CAJ, Di Santo JP. Developmental options and functional plasticity of innate lymphoid cells. Curr. Opin. Immunol. 2017;44:61–68
Eberl G, Colonna M, Di Santo JP, McKenzie ANJ. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:6566
Zhang J, Marotel M, Fauteux-Daniel S et al. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur J Immunol. 2018;48:738–750
Poggi A, Benelli R, Vene R et al. Human gut-associated natural killer cells in health and disease. Front Immunol. 2019;10:961
Bernink JH, Peters CP, Munneke M et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol. 2013;14:221–229
Fuchs A, Vermi W, Lee JS et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity. 2013;38:769–781
Hwang YY, McKenzie AN. Innate lymphoid cells in immunity and disease. Adv Exp Med Biol. 2013;785:9–26
Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol. 2019;105:1319–1329
Campos TM, Novais FO, Saldanha M, et al. Granzyme B produced by natural killer cells enhances inflammatory response and contributes to the immunopathology of cutaneous leishmaniasis. J Infect Dis. 2019.
Klein Wolterink RG, Serafini N, van Nimwegen M et al. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc Natl Acad Sci USA 2013;110:10240–10245
Hoyler T, Klose Christoph SN, Souabni A et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37:634–648
Spooner CJ, Lesch J, Yan D et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol. 2013;14:1229–1236
Yu Y, Wang C, Clare S et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J Exp Med. 2015;212:865–874
Kim BS, Wojno ED, Artis D. Innate lymphoid cells and allergic inflammation. Curr Opin Immunol. 2013;25:738–744
Jiang M, Tao S, Zhang S et al. Type 2 innate lymphoid cells participate in IL-33-stimulated Th2-associated immune response in chronic obstructive pulmonary disease. Exp Ther Med. 2019;18:3109–3116
Helfrich S, Mindt BC, Fritz JH, Duerr CU. Group 2 innate lymphoid cells in respiratory allergic inflammation. Front Immunol. 2019;10:930
Herbert DR, Douglas B, Zullo K. Group 2 innate lymphoid cells (ILC2): type 2 immunity and helminth immunity. Int J Mol Sci. 2019;20:2276
Rafei-Shamsabadi DA, Klose CSN, Halim TYF, Tanriver Y, Jakob T. Context dependent role of type 2 innate lymphoid cells in allergic skin inflammation. Front Immunol. 2019;10:2591
Qiu J, Heller JJ, Guo X et al. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity. 2012;36:92–104
Britanova L, Diefenbach A. Interplay of innate lymphoid cells and the microbiota. Immunol Rev. 2017;279:36–51
Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, Cyster JG. IgA production requires B cell interaction with subepithelial dendritic cells in Peyers patches. Science. 2016;352:4822
Kruglov AA, Grivennikov SI, Kuprash DV et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science. 2013;342:1243–1246
Takatori H, Kanno Y, Watford WT et al. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med. 2009;206:35–41
Klose CS, Kiss EA, Schwierzeck V et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature. 2013;494:261–265
Mackley EC, Houston S, Marriott CL et al. CCR7-dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat Commun. 2015;6:5862
Song C, Lee JS, Gilfillan S et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J Exp Med. 2015;212:1869–1882
Zeng B, Shi S, Ashworth G, Dong C, Liu J, Xing F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Disease. 2019;10:315
Powell N, Lo JW, Biancheri P et al. Interleukin 6 increases production of cytokines by colonic innate lymphoid cells in mice and patients with chronic intestinal inflammation. Gastroenterology. 2015;149:e15
Pearson C, Thornton EE, McKenzie B et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife. 2016;5:e10066
Giacomin PR, Moy RH, Noti M et al. Epithelial-intrinsic IKKalpha expression regulates group 3 innate lymphoid cell responses and antibacterial immunity. J Exp Med. 2015;212:1513–1528
Aparicio-Domingo P, Romera-Hernandez M, Karrich JJ et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J Exp Med. 2015;212:1783–1791
Pantazi E, Powell N. Group 3 ILCs: peacekeepers or troublemakers? What’s Your Gut Telling You?! Front Immunol. 2019;10:676
Gronke K, Hernandez PP, Zimmermann J et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. 2019;566:249–253
Geremia A, Arancibia-Carcamo CV, Fleming MP et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. 2011;208:1127–1133
Diefenbach A, Colonna M, Koyasu S. Development, differentiation, and diversity of innate lymphoid cells. Immunity. 2014;41:354–365
Bernink Jochem H, Krabbendam L, Germar K et al. Interleukin-12 and -23 control plasticity of CD127+ Group 1 and Group 3 innate lymphoid cells in the intestinal lamina propria. Immunity. 2015;43:146–160
Teunissen MBM, Munneke JM, Bernink JH et al. Composition of innate lymphoid cell subsets in the human skin: enrichment of NCR + ILC3 in lesional skin and blood of psoriasis patients. J Invest Dermatol. 2014;134:2351–2360
Viant C, Rankin LC, Girard-Madoux MJ, et al. Transforming growth factor-beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci Signal. 2016;9:ra46.
Chea S, Perchet T, Petit M, et al. Notch signaling in group 3 innate lymphoid cells modulates their plasticity. Sci Signal. 2016;9:ra45.
Ebbo M, Crinier A, Vely F, Vivier E. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol. 2017;17:665–678
Lim AI, Menegatti S, Bustamante J et al. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J Exp Med. 2016;213:569–583
Li S, Bostick JW, Ye J et al. Aryl Hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity. 2018;49:915
Forkel M, van Tol S, Hoog C, Michaelsson J, Almer S, Mjosberg J. Distinct Alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established crohn’s disease and ulcerative colitis. J Crohns Colitis. 2019;13:67–78
Kinnebrew MA, Buffie CG, Diehl GE et al. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity. 2012;36:276–287
Goto Y, Obata T, Kunisawa J et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science. 2014;345:1254009
Tsai PY, Zhang B, He WQ et al. IL-22 upregulates epithelial claudin-2 to drive diarrhea and enteric pathogen clearance. Cell Host Microbe. 2017;21:e4
Kobori A, Yagi Y, Imaeda H et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol. 2010;45:999–1007
Schwartz C, O’Grady K, Lavelle EC, Fallon PG. Interleukin 33: an innate alarm for adaptive responses beyond Th2 immunity-emerging roles in obesity, intestinal inflammation, and cancer. Eur J Immunol. 2016;46:1091–1100
Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci USA 2015;112:10762–10767
Schiering C, Krausgruber T, Chomka A et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568
Forkel M, van Tol S, Höög C, Michaëlsson J, Almer S, Mjösberg J. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established crohn’s disease and ulcerative colitis. J Crohns Colitis. 2019;13:67–78
Camelo A, Barlow JL, Drynan LF et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J Gastroenterol. 2012;47:1198–1211
Zelante T, Iannitti RG, Cunha C et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity. 2013;39:372–385
Qiu J, Guo X, Chen ZM et al. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–399
Ganal-Vonarburg SC, Duerr CU. The interaction of intestinal microbiota and innate lymphoid cells in health and disease throughout life. Immunology. 2020;159:39–51
Constantinides MG. Interactions between the microbiota and innate and innate-like lymphocytes. J Leukoc Biol. 2018;103:409–419
Blander JM, Longman RS, Iliev ID, Sonnenberg GF, Artis D. Regulation of inflammation by microbiota interactions with the host. Nat Immunol. 2017;18:851–860
Thaiss CA, Zmora N, Levy M, Elinav E. The microbiome and innate immunity. Nature. 2016;535:65–74
Sawa S, Lochner M, Satoh-Takayama N et al. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326
Sonnenberg GF, Monticelli LA, Alenghat T et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325
Penny HA, Hodge SH, Hepworth MR. Orchestration of intestinal homeostasis and tolerance by group 3 innate lymphoid cells. Semin Immunopathol. 2018;40:357–370
Castleman MJ, Dillon SM, Purba CM et al. Commensal and pathogenic bacteria indirectly induce IL-22 but Not IFNgamma production from human colonic ILC3s via multiple mechanisms. Front Immunol. 2019;10:649
Babu ST, Niu X, Raetz M, Savani RC, Hooper LV, Mirpuri J. Maternal high-fat diet results in microbiota-dependent expansion of ILC3s in mice offspring. JCI Insight. 2018;3.
Zhou L, Chu C, Teng F et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature. 2019;568:405–409
Wang Y, Kuang Z, Yu X, Ruhn KA, Kubo M, Hooper LV. The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science. 2017;357:912–916
Godinho-Silva C, Domingues RG, Rendas M, et al. Light-entrained and brain-tuned circadian circuits regulate ILC3s and gut homeostasis. Nature. 2019.
Teng F, Goc J, Zhou L, et al. A circadian clock is essential for homeostasis of group 3 innate lymphoid cells in the gut. Sci Immunol. 2019;4.
von Moltke J, Ji M, Liang HE, Locksley RM. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. 2016;529:221–225
Frisbee AL, Saleh MM, Young MK et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat Commun. 2019;10:2712
Garrido-Mesa N, Schroeder JH, Stolarczyk E et al. T-bet controls intestinal mucosa immune responses via repression of type 2 innate lymphoid cell function. Mucosal Immunol. 2019;12:51–63
Sepahi A, Liu Q, Friesen L, Kim CH. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 2020.
Lloyd-Price J, Arze C, Ananthakrishnan AN et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662
Lamas B, Richard ML, Leducq V et al. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med. 2016;22:598–605
Scott SA, Fu J, Chang PV. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc Natl Acad Sci USA 2020;117:19376–19387
Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301
Sano T, Huang W, Hall JA et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid A to promote local effector Th17 responses. Cell. 2015;163:381–393
Atarashi K, Tanoue T, Ando M et al. Th17 cell induction by adhesion of microbes to intestinal epithelial cells. Cell. 2015;163:367–380
Mielke LA, Jones SA, Raverdeau M et al. Retinoic acid expression associates with enhanced IL-22 production by gammadelta T cells and innate lymphoid cells and attenuation of intestinal inflammation. J Exp Med. 2013;210:1117–1124
Longman RS, Diehl GE, Victorio DA et al. CX(3)CR1(+) mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J Exp Med. 2014;211:1571–1583
Castellanos JG, Woo V, Viladomiu M et al. Microbiota-induced TNF-like ligand 1A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity. 2018;49:e5
Mortha A, Chudnovskiy A, Hashimoto D et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288
Levy M, Thaiss CA, Zeevi D et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell. 2015;163:1428–1443
Munoz M, Eidenschenk C, Ota N et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity. 2015;42:321–331
Hepworth MR, Fung TC, Masur SH et al. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science. 2015;348:1031–1035
Hepworth MR, Monticelli LA, Fung TC et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature. 2013;498:113–117
Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14:269–278
Withers DR, Hepworth MR, Wang X et al. Transient inhibition of ROR-gammat therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat Med. 2016;22:319–323
Hueber W, Sands BE, Lewitzky S et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700
Withers DR, Hepworth MR. Group 3 innate lymphoid cells: communications hubs of the intestinal immune system. Front Immunol. 2017;8:1298
Kaser A. Not all monoclonals are created equal-lessons from failed drug trials in Crohn’s disease. Best Pract Res Clin Gastroenterol. 2014;28:437–449
Colombel JF, Sendid B, Jouault T, Poulain D. Secukinumab failure in Crohn’s disease: the yeast connection? Gut. 2013;62:800–801
Zhou L, Sonnenberg GF. Essential immunologic orchestrators of intestinal homeostasis. Sci Immunol. 2018;3.
Sandborn WJ, Ferrante M, Bhandari BR, et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with ulcerative colitis. Gastroenterology. 2019.
Geremia A, Arancibia-Carcamo CV. Innate lymphoid cells in intestinal inflammation. Front Immunol. 2017;8:1296
Tang C, Kakuta S, Shimizu K, et al. Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat Immunol. 2018.
Reinisch W, de Villiers W, Bene L et al. Fontolizumab in moderate to severe Crohn’s disease: a phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm Bowel Dis. 2010;16:233–242
Goldberg R, Prescott N, Lord GM, MacDonald TT, Powell N. The unusual suspects–innate lymphoid cells as novel therapeutic targets in IBD. Nat Rev Gastroenterol Hepatol. 2015;12:271–283
Rutgeerts P, Sandborn WJ, Feagan BG et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476
Pelczar P, Witkowski M, Perez LG et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science. 2016;354:358–362
Danese S, Vermeire S, Hellstern P et al. Randomised trial and open-label extension study of an anti-interleukin-6 antibody in Crohn’s disease (ANDANTE I and II). Gut. 2019;68:40–48
Powell N, Walker AW, Stolarczyk E et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity. 2012;37:674–684
Ermann J, Staton T, Glickman JN, de Waal Malefyt R, Glimcher LH. Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice. Proc Natl Acad Sci USA. 2014;111:E2559-66.
Acknowledgment
This manuscript preparation was supported by the National Natural Science Foundation of China (grant no. 81970494 and 81670504) and Key Research and Development Program of Hunan Province (grant no. 2019SK2041).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Luo, W., Tian, L., Tan, B. et al. Update: Innate Lymphoid Cells in Inflammatory Bowel Disease. Dig Dis Sci 67, 56–66 (2022). https://doi.org/10.1007/s10620-021-06831-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-021-06831-8