
Abstract
Background
Oxidative stress has been suggested to be a factor contributing to the disease severity of inflammatory bowel disease (IBD). BJ-1108, a derivative of 6-amino-2,4,5-trimethylpyridin-3-ol, is reported to significantly inhibit the generation of reactive oxygen species (ROS) in vitro. However, whether this molecule affects intestinal inflammation is largely unknown. We aimed to investigate the effect of BJ-1108 on dextran sulfate sodium (DSS)-induced experimental colitis in mice.
Methods
Colitis was induced in mice with DSS, and disease severity was estimated by evaluating body weight, colon length, histology, immune cell infiltration, and intestinal permeability. We examined the protective effects of BJ-1108 on barrier function using Caco-2 cells. Last, we estimated the impact of BJ-1108 on the phosphorylation of NF-kB, PI3K/AKT, and mitogen-activated protein kinases.
Results
Mice treated with BJ-1108 exhibited improved disease severity, as indicated by evaluations of body weight, histological scores, spleen weight, and infiltrates of T cells and macrophages. The administration of BJ-1108 inhibited the colonic mRNA expression of IL-6 and IL-1β in vivo. Additionally, BJ-1108 limited intestinal permeability and enhanced the expression of tight junction (TJ) proteins such as claudin-1 and claudin-3 in the DSS-induced colitis model. In an in vitro model using Caco-2 cells, BJ-1108 ameliorated cytokine-induced ROS generation in a dose-dependent manner and remarkably recovered barrier dysfunction as estimated by evaluating transepithelial electrical resistance and TJ protein expression. BJ-1108 suppressed the NF-kB/ERK/PI3K pathway.
Conclusions
This study demonstrated that BJ-1108 ameliorated intestinal inflammation in an experimental colitis mouse model, suggesting possible therapeutic implications for IBD.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammatory bowel disease (IBD) is a chronic inflammatory condition of the intestinal wall in humans. Although the exact etiology of IBD is not fully understood, various factors have been suggested to be the cause of IBD, including abnormal immune responses, genetics, intestinal microflora, and environmental circumstances [1]. The traditional therapies for patients with IBD mainly consist of anti-inflammatory and immunomodulatory treatments. Additionally, biologic therapies have been introduced and widely used in the treatment of IBD. However, the systemic administration of these agents has limitations due to infection, tumorigenesis, intolerance, and high cost. Therefore, the development of new therapeutic agents is strongly needed.
Reactive oxygen species (ROS) are one of the risk factors contributing to the development of IBD [2, 3]. Under normal conditions, intestinal epithelial cells (IECs) maintain their scaffold by adhering with neighboring cells via tight junctional (TJ) proteins, supporting intestinal homeostasis against various foreign antigens. When mucosal inflammation occurs, ROS are generated by IECs, neutrophils, and macrophages [3]. In turn, excessive ROS result in alterations in TJs and consequently lead to disruption of intestinal barrier function. There are some reports that patients with IBD have impaired expression of the oxidase enzymes involved in ROS generation (e.g., NOX and myeloperoxidase, MPO) and intracellular antioxidants (e.g., SOD and GSH) [4,5,6].
BJ-1108 is a 6-amino-2,4,5-trimethylpyridin-3-ol analogue that was previously suggested to have antioxidant and antiangiogenic activity in vitro [7, 8]. A study reported that BJ-1108 treatment significantly diminished 5-hydroxytryptamine (5-HT)-induced angiogenesis, tumor proliferation, and ROS generation [9]. Additionally, BJ-1108 significantly inhibits IFN-γ- and IL-17α-producing cells in ovalbumin-challenged mice and attenuates experimental autoimmune encephalomyelitis in myelin oligodendrocyte glycoprotein-induced mice [10]. However, the anticolitogenic effect of BJ-1108 is largely unknown.
The dextran sulfate sodium (DSS)-induced colitis model established in mice is regarded as a relevant animal model. Previously, it was demonstrated that DSS treatment induces significantly elevated expression of ROS through the nuclear factor-kB (NF-kB) pathway in a human colonic epithelial cell line, which can be rescued by treatment with a superoxide scavenger [11]. Therefore, the aims of this study were to determine the anticolitogenic effects of BJ-1108 using an in vivo DSS-induced colitis model and investigate the possible signaling pathway by using an in vitro Caco-2 system.
Materials and Methods
Experimental Colitis Induced by DSS in Mice and Colitis Evaluation
Wild-type male C57BL/6N mice (8 weeks old, n = 8) were maintained under specific pathogen-free conditions at 22 °C with a 12-h light/dark cycle. To induce colitis, animals were administered 2% DSS (MP Biomedicals, USA) in the drinking water for 7 days, followed by housing with normal drinking water for 14 days. Then, the 2% DSS administration in the drinking water was repeated for 7 days, followed by housing with normal drinking water for 7 days. The control group received normal drinking water continuously. Mice were orally administered either BJ-1108 (1 or 3 mg/kg) or a vehicle (corn oil). All mice were fasted overnight and killed. The severity of colitis was investigated by evaluating colon length, histology, spleen size, in vivo intestinal permeability, and colonic MPO activity.
Histological scores were evaluated using previously published scoring systems [12]. To measure in vivo intestinal permeability, 4-kDa fluorescein isothiocyanate dextran (FITC-dextran) (Sigma-Aldrich, Seoul, Korea) was administered to mice by oral gavage. After 4 h, the serum FITC-dextran level was measured using a fluorescence spectrometer with a standard. MPO activity was measured by assessing an enzymatic reaction in the presence of peroxide and o-dianisidine (D3252, Sigma) [13]. Briefly, colon tissue samples were homogenized in a hexadecyltrimethylammonium bromide buffer (H5882, Sigma) and placed in a 96-well microplate. The enzymatic reaction was initiated by adding a peroxide and o-dianisidine solution. The absorbance was read at 450 nm.
This study was approved by the animal care committee of Kyungpook National University, and all experiments were performed in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care International.
Immunohistochemical (IHC) Staining
Paraffin-embedded sections were rehydrated, and endogenous peroxidase was blocked by treatment with H2O2 for 15 min. Epitope retrieval was performed in Tris–EDTA buffer or sodium citrate buffer. The slides were blocked with 5% bovine serum albumin and then incubated with either an anti-CD3 (A0452, Agilent) or anti-F4/80 (MCA497GA, Bio-Rad) antibody for 30 min. Labeling was visualized with 3,3′-diaminobenzidine (DAB)(K3468, Dako).
Quantitative PCR (qPCR)
Total RNA was extracted from homogenized colon tissue with Qiazol (Qiagen) and purified with a Qiagen RNeasy mini isolation kit (Qiagen). RNA purity was measured with a Nanodrop (Thermo Fisher). A reverse transcription prime kit (EBT-1520, ELPIS Biotech) was used to generate a cDNA library. qPCR was performed according to the manufacturer’s recommendation with PowerUp SYBR Green Master Mix (4367659, Thermo Fisher). Data were analyzed with Vii™ 7 Real-Time PCR. The primer sequences were as follows: IL-12b (forward, 5′-AGA-CCC-TGC-CCA-TTG-AAC-TG-3′ and reverse, GAA-GCT-GGT-GCT-GTA-GTT-CTC-ATA-TTT), Tjp1 (forward, 5′-TCA-TCC-CAA-ATA-AGA-ACA-GAG-C-3′and reverse, 5′-GAA-GAA-CAA-CCC-TTT-CAT-AAG-C-3′), Ocln (forward, 5′-AAG-CAA-GTG-AAG-GGA-TCT-GC-3′and reverse, 5′-GGG-GTT-ATG-GTC-CAA-AGT-CA-3′), Cldn1 (forward, 5′-GCT-GTG-CTG-CTA-ACC-CTG-TTG-3′ and reverse, 5′-TGC-AGG-ATC-TGG-GAT-GGA-AA-3′), Cldn3 (forward, 5′-CCA-CCC-CAC-CTT-CCA-GAT-G-3′ and reverse, 5′-GGA-GAT-GCC-AGC-CTG-TCT-GT-3′), Cldn4 (forward, 5′-GTG-TCC-TGG-ACC-GCT-CAC-A-3′ and reverse, 5′-GCC-CCC-ATT-TCC-CTC-TTC-T-3′), IL-1b (forward, 5′-TTC-AGG-CAG-GCA-GTA-TCA-CTC-3′ and reverse, 5′-GAA-GGT-CCA-CGG-GAA-AGA-CAC-3′), IL-6 (forward, 5′-TTC-TCT-GGG-AAA-TCG-TGG-AAA-3′ and reverse, 5′-TGC-AAG-TGC-ATC-ATC-GTT-GTT-3′), and IL-10 (forward, 5′-CAG-AGC-CAC-ATG-CTC-CTA-GA-3′ and reverse, 5′-GTC-CAG-CTG-GTC-CTT-TGT-TT-3′).
ROS Detection in Caco-2 Cells
Intracellular ROS were detected with a dihydroethidium (DHE) fluorescent probe and quantified by fluorescence confocal microscopy and florescence photospectroscopy. Briefly, a monolayer of Caco-2 cells was incubated with 1 µM DHE (D7008, Sigma) for 15 min at 37 °C, washed and fixed with 10% formalin. The nuclear counterstain 4′,6-diamidino-2-phenylindole (DAPI) was added for confocal microscopy.
Western Blot Analysis
A Western blot assay was performed by using antibodies specific for p65 (Cell Signal Technology, #8242), p-p65 (CST, #3033), p38 mitogen-activated protein kinase (MAPK) (CST, #9212), p-p38 MAPK (CST, #4631), phosphoinositide 3-kinase kinase (PI3K) p85 (19H8), p-PI3K p85 (CST, #4257), extracellular signal regulated kinase (ERK) (BD, 610123), p-ERK (CST, #9101), c-Jun N-terminal kinase 2 (JNK2) (56G8) (CST, #9258), phosphor stress-activated protein kinase/c-Jun NH2-terminal kinase (p-SAPK/JNK) (Thr183/Tyr185)(81E11) (CST, #4668), protein kinase B (AKT) (CST, #9272), p-AKT (ser473) (CST, #9271), mammalian target of rapamycin (mTOR) (CST, #2972), and p-mTOR (CST, #2972).
Cell Viability Assay
Drug cytotoxicity in Caco-2 cells was examined by monitoring extracellular lactate dehydrogenase (LDH) using a CytoTox96® non-radioactive cytotoxicity assay kit (G1780, Promega). The levels of released LDH in culture supernatants were measured with a 30-min coupled enzymatic assay according to the manufacturer’s instructions.
Transepithelial Electrical Resistance (TEER)
Approximately 1.5 × 105 Caco-2 cells were cultured on the collagen-coated transwell insert (CLS3460, Sigma) with 200 μl culture medium added to the apical chamber and 1000 μl to the basolateral chamber for two weeks before TEER measurement. The epithelial voltmeter EVOM STX2 (World Precision Instruments) was used. Resistance was defined according to the surface of the transwell filters (Ω × cm2), and the TEER of cell-free inserts was calculated from these values.
Statistical Analysis
Statistical analysis was performed using Excel (Microsoft, USA) or GraphPad Prism5 (La Jolla, CA, USA).
Results
Treatment with BJ-1108 Ameliorated Experimental Colitis Induced by DSS in Mice
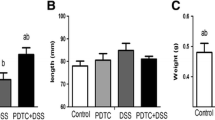
To determine the anticolitogenic effects of the BJ-1108 compound, we used an experimental colitis mouse model induced by DSS, which is known to disrupt ROS homeostasis in IECs and consequently lead to defective TJ integrity, cell cycle arrest, and apoptosis [14, 15]. It was found that two cycles of DSS administration in mice successfully induced severe inflammation in the colon, which was indicated by weight loss, colon length reduction, spleen swelling, high histological scores, elevated MPO activity, and increased intestinal permeability estimated with FITC-dextran (Fig. 1).

BJ-1108 treatment ameliorates experimental DSS-induced colitis in mice. C57BL/6N mice were treated with 2% DSS in the drinking water for 1 week, followed by housing with normal drinking water for 2 weeks, and then 2% DSS in the drinking water for 1 week, followed by housing with normal drinking water for 1 week. BJ-1108 compounds (1 or 3 g/kg) were administered daily via oral gavage. a Weight changes of control, vehicle control (DSS), and BJ-1108-treated mice during DSS-induced colitis. b Representative gross image of the colon. c Mean colon lengths. d, e Representative H&E staining of colon samples with histological severity scores. Scale bar size 100 μm. f MPO activity. g Plasma FITC-dextran 4-kDa level
Next, we administered BJ-1108 daily by oral gavage during the DSS cycles and examined inflammation severity in mice. Interestingly, the mice administered BJ-1108 appeared to have enhanced weight recovery, especially after 24 days of DSS treatment, compared with the control mice (Fig. 1a). Moreover, histological examinations revealed that BJ-1108 treatment significantly limited tissue damage (Fig. 1d, e). BJ-1108 administration significantly mitigated serum FITC-dextran levels in the treated mice compared with the control mice (Fig. 1g), indicating decreased gut permeability.
Last, BJ-1108 tended to attenuate colon length shortening and MPO activity, but these differences were not significant (Fig. 1c, f). The weight of the enlarged spleen was significantly recovered in the BJ-1108-treated group compared with the DSS control group (Supplementary Figure 1).
The infiltration of T cells and macrophages in the colon was determined by IHC staining (Fig. 2). The numbers of CD3+ T cells in the mucosal area were remarkably reduced in the BJ-1108-treated group compared with the control group (Fig. 2a, b). Moreover, the numbers of infiltrated F4/80+ macrophages in the intestinal mucosa were reduced after BJ-1108 administration (Fig. 2c, d). These data suggest that treatment with BJ-1108 has anticolitogenic effects on the experimental DSS-induced colitis model.

BJ-1108 treatment reduced colonic CD3+ T cell and F4/80+ macrophage numbers. a Representative IHC images of CD3 expression in the colon of control, vehicle control (DSS), and BJ-1108-treated mice (1 or 3 mg/kg). b Relative intensity of CD3+ DAB chromogen (per section). c Representative IHC images of F4/80 expression in the colon of control, vehicle control, and BJ-1108-treated mice. d Relative intensity of F4/80+ DAB chromogen (per section). Scale bar size 100 μm
BJ-1108 Downregulated IL-1β, IL-6, IL-12, and IL-10 Expression in the Colon
Next, we asked whether BJ-1108 treatment mitigates the mRNA expression of various inflammatory cytokines such as IL-1β, IL-6, IL-12, and IL-10, which were previously shown to exhibit highly upregulated expression in DSS-induced colitis mice [16]. Mice fed 2% DSS expressed higher IL-1β, IL-6, IL-12, and IL-10 mRNA levels in the colon than did control mice (Fig. 3a–d). In contrast, BJ-1108-treated mice exhibited significant reductions in IL-1β, IL-6, IL-12, and IL-10 transcription (Fig. 3a–d). This overall downregulation of cytokine expression possibly reflected the amelioration of DSS-induced colitis mediated by BJ-1108, as shown in Figs. 1 and 2.

BJ-1108 treatment compromised cytokines in response to DSS injury. The mRNA expression of cytokines such as a IL-1b, b IL-6, c IL-10, and d IL-12b in the colon of control, vehicle control (DSS), and BJ-1108-treated mice (1 or 3 mg/kg) was evaluated by qPCR. All the mRNA levels were normalized to the glyceraldehyde-3-phosphate dehydrogenase (Gapdh) level
BJ-1108 Ameliorated Intestinal Permeability by Upregulating TJ Protein Expression
Impaired expression of TJ proteins is related to altered intestinal epithelial integrity and increased intestinal permeability in colitis [17, 18]. In this study, we investigated TJ protein expression in the colon and in vivo intestinal permeability. The mRNA expression of Tjp1 and Ocln (encoding the ZO-1 and occludin proteins, respectively) in the colon was significantly downregulated after DSS treatment compared with control treatment, whereas mice administered BJ-1108 exhibited significantly higher levels of mRNA expression of Tjp1 and Ocln (Fig. 4a, b). However, the protein expression of ZO-1 and occludin was not influenced by BJ-1108 treatment (Fig. 4c–e). Nonetheless, the levels of other TJ proteins such as claudin-1, claudin-3, and claudin-4 were restored after BJ-1108 administration in DSS-treated mice (Fig. 4c, f–h).

BJ-1108 treatment rescued altered TJ protein expression. The mRNA expression of Tjp1 (a) and Ocln (b) in the colon of control, vehicle control (DSS), and BJ-1108-treated mice (1 or 3 mg/kg) was determined by qPCR and normalized to the glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNA expression. c The protein expression of ZO-1, occludin, claudin-1, claudin-3, and claudin-4 in the colon was determined by Western blotting and quantified relative to β-actin protein expression (e–h)
BJ-1108 Enhanced Intestinal Barrier Functions and Reduced ROS Production In Vitro
To further determine the protective role of BJ-1108 compounds in intestinal barrier function, we used an in vitro system consisting of a human colonic epithelial cell line (Caco-2 cells). A monolayer of Caco-2 cells grown for 2 weeks was exposed to TNF-α and IFN-γ, which are major cytokines that have been shown to induce oxidative stress in IECs in a colitis animal model and the rearrangement of TJ molecules in vitro [19,20,21]. Furthermore, the previous study has been shown that the combination of IFN-γ and TNF-α has superior effects on deforming tight junctional structure and inducing secretion of cytokines (IL-6 and IL-8) [21,22,23]. The function of the barrier was documented by visualization of TJs by confocal immunofluorescence (IF) imaging for ZO-1 and TEER measurement. Indeed, treatment with TNF-α/IFN-γ increased the tortuosity and rearrangement of the ZO-1 TJ scaffold (Fig. 5a) and significantly disrupted TEER (Fig. 5b). BJ-1108 treatment markedly ameliorated these altered barrier functions (Fig. 5a, b).

Injured intestinal barrier function and intracellular ROS levels in Caco-2 cells were rescued after BJ-1108 treatment. a Representative IF images of ZO-1 (green) and the nucleus (blue) in Caco-2 cells treated with BJ-1108 (1 or 10 μM) in the absence or presence of TNF-α and IFN-γ (each 40 ng/mL). BJ-1108 reduced reactive oxygen species (ROS) production in vitro. Scale bar size 30 μm for ×200 magnification and 10 μm for ×600 magnification. b Percentage change in TEER after 24 h. Monolayers of Caco-2 cells were treated with BJ-1108 (1 or 10 μM) in the absence or presence of TNF-α (40 ng/mL) and IFN-γ (40 ng/mL) for 24 h. c Representative IF images of DHE (red) and DAPI (blue). Scale bar size 30 μm. d Relative intensity of DHE (per section). e Relative intensity levels of DHE in experimental samples compared to untreated control samples
In addition, ROS production was measured by DHE staining to investigate the antioxidant effects of BJ-1108. Treatment with TNF-α/IFN-γ greatly increased ROS production in Caco-2 cells compared with no treatment (Fig. 5c–e). The addition of BJ-1108 significantly reduced ROS generation in Caco-2 cells in a dose-dependent manner (Fig. 5c–e) without causing necrosis, as indicated by monitoring LDH release (Supplementary Figure 2).
BJ-1108 Decreased NF-κB and PI3K/AKT Expression and Increased ERK Expression in Caco-2 Cells
After the determination of the enhanced intestinal barrier function and antioxidant effects of BJ-1108 on Caco-2 cells, we further examined the potential signaling pathway through which BJ-1108 might mediate the protective effects against ROS. To do this, we incubated Caco-2 cells with BJ-1108 and then TNF-α/IFN-γ.
First, we evaluated the expression of the inflammatory transcription factor NF-κB p65 and its phosphorylation by Western blotting. Pretreatment with BJ-1108 significantly downregulated p-p65 expression (Fig. 6a). Incubation with TNF-α/IFN-γ increased the p-p65/p65 ratio in the absence of BJ-1108, whereas the p-p65/p65 ratio was significantly reduced in the presence of BJ-1108 (Fig. 6b), suggesting that the anti-inflammatory effects of BJ-1108 were mediated by NF-κB dephosphorylation.

BJ-1108 treatment affects NF-κB, JNK and AKT phosphorylation in Caco-2 cells. a Western blot analysis of p-p65 and total p65 in Caco-2 cells pretreated with BJ-1108 (10 μM) for 30 min in the absence or presence of TNF-α (40 ng/mL) and IFN-γ (40 ng/mL) for 5, 15, 30, 60, or 120 min. b Relative intensity of p-p65 levels to total p65 levels. c Western blot analysis of p-ERK, total ERK, p-p38, total p38, p-JNK and total JNK in Caco-2 cells pretreated with BJ-1108 (10 μM) for 30 min in the absence or presence of TNF-α (40 ng/mL) and IFN-γ (40 ng/mL) for 5, 15, 30, 60, or 120 min. d–g Relative intensity of p-ERK, p-p38, and p-JNK levels to the respective total form level. h Western blot analysis of p-AKT, total AKT, p-p85, total p85, p-mTOR, and mTOR in Caco-2 cells pretreated with BJ-1108 (10 μM) for 30 min in the absence or presence of TNF-α (40 ng/mL) and IFN-γ (40 ng/mL) for 5, 15, 30, 60, or 120 min. i–k Relative intensity of p-AKT, p-p85, and p-mTOR levels compared to the respective total form level
Next, we examined whether the anticolitogenic effects of BJ-1108 were possibly mediated by the MAPK signaling pathway in Caco-2 cells. The results showed that Caco-2 cells pretreated with BJ-1108 significantly increased p-ERK expression, while total ERK1/2 expression remained constant without stimulation (Fig. 6c). After treatment with TNF-α/IFN-γ, the vehicle control had a tendency to increase p-ERK expression in a time-dependent manner, whereas the drug-treated group sustained a high level of p-ERK expression (Fig. 6d). However, this difference was not significant. For MAPK signaling pathway molecules, we also examined the impact of BJ-1108 on p38 and JNK expression by Western blotting (Fig. 6c). The results suggested that the anticolitogenic action of BJ-1108 was independent of the p38 and JNK signaling pathways (Fig. 6e, f).
Finally, we examined whether BJ-1108 treatment affects the AKT/PI3K/mTOR axis in Caco-2 cells. The Western blot image shows a significant reduction in the expression of p-PI3K in Caco-2 cells treated with BJ-1108 compared with the corresponding control cells (Fig. 6h, j). However, the expression of AKT and p-AKT was not affected by BJ-1108 treatment. In addition, p-mTOR expression was significantly downregulated by pretreatment with BJ-1108 (Fig. 6k). Taken together, these results suggest that the anticolitogenic effect of BJ-1108 on injured IECs is mediated mainly through NF-κB and PI3K inhibition.
Discussion
This in vivo and in vitro study revealed that BJ-1108 significantly improved intestinal inflammation by reducing ROS production, decreasing cytokine levels, and enhancing intestinal barrier functions. The NF-kB and PI3K pathway might be involved in the anti-inflammatory effect of BJ-1108 on the colitis model.
Antioxidant drugs have been investigated to evaluate their therapeutic efficacy in IBD using various experimental colitis models. N-acetylcysteine (NAC), a synthetic antioxidant, significantly attenuates the development of 2,4,6-trinitrobenzenesulfonic acid (TNBS)-induced colitis in rats, as indicated by MPO activity measurement and histological examination, in comparison with mesalamine alone [24]. NAC reduced colonic COX-2 transcript expression, PGE-2 protein expression, and inducible nitric oxide synthase activity in TNBS-treated rats [25]. The protective effects of NAC have also been demonstrated in a DSS-induced colitis mouse model by ameliorating histological scores, MPO activity, cytokine levels, and ROS generation [26]. NAC administration prevents epithelial cell apoptosis, necrosis, oxidative stress, mucin depletion, and MPO activity in DSS-treated mice [27]. These results support our findings that BJ-1108, a derivative of an antioxidant, successfully lowered histological scores, MPO levels, cytokine production, and immune cell infiltration in the experimental DSS-induced colitis model in vivo.
Antioxidant therapy has been shown to modulate the function of the mucosal barrier in vivo and in vitro. Treatment with berberine, which is known to reduce ROS production [28], significantly diminishes oxidative stress in the colon and improves intestinal barrier function in DSS-induced colitis mice compared with control mice [29]. Additionally, treatment with quercetin and antioxidants remarkably restores ROS-injured paracellular permeability and TJ protein redistribution in Caco-2 cells [30, 31]. In the present study, we confirmed that BJ-1108 significantly ameliorated intestinal barrier dysfunction, as evidenced by reduced FITC-dextran absorption and preserved TJ protein expression in DSS-treated mice in vivo, as well as attenuated ROS production, reduced paracellular permeability, and TJ redistribution in vitro (Fig. 5).
In the in vitro model, we noticed that treatment with BJ-1108 decreased NF-kB phosphorylation. The NF-kB signaling pathway plays a critical role in intestinal barrier homeostasis, which may be altered in patients with IBD. In humans, the activation and expression of NF-kB in the mucosa of IBD patients are reported to have positive correlations with the severity of intestinal inflammation [32]. In an animal study, knocking down the expression of p65, a subunit of NF-kB, significantly attenuated disease severity in TNBS-induced or IL-10 knockout colitis mouse models [33]. Additionally, conditionally knocking out IKKβ, an NF-kB regulator, suppresses acute colitis in a DSS-induced colitis model [34]. Moreover, treatment with telmisartan, an antioxidant/anti-inflammatory agent targeting the angiotensin II receptor, significantly attenuates the expression of NF-κB and MPO and oxidative stress in the colonic tissues of rats with TNBS-induced colitis [35].
The detailed mechanisms underlying the protective role of NF-kB dephosphorylation by BJ-1108 are unclear. Activation of NF-κB seems to be involved in pro-oxidant gene expression such as NADPH oxidase organizer 1 (NOXO1), one of the components forming NADPH oxidase 1 (NOX1) complex which is known to produce ROS in epithelial cells [36]. Notably, the inhibition of p65, a subunit NF-κB, expression markedly suppressed the TNF-α-induced noxo1 gene transcription [36]. Although we did not perform experiment to evaluate NOX1 expression in this study, we surmise that antioxidant effect of BJ-1108 may be possibly mediated by reduced nox1 gene expression by NF-kB dephosphorylation at least in part.
We found that BJ-1108 increased ERK expression in vitro. Data of ERK effects on the tight junction of epithelial cells are conflicting in the literature. Some reports showed that intracellular ERK phosphorylation have a negative relationship with the intestinal epithelial cell differentiation process. ERK 1/2 phosphorylation markedly reduced in differentiated Caco-2/15 cells after 12 days post-confluent [37]. Furthermore, inhibition of ERK1/2 phosphorylation by AG1478 or U0125 inhibitors completely prevented cytokine-induced TJ disassembly [38]. On the other hand, some studies showed that ERK activation can protect the disruption of barrier integrity. Hydrogen peroxide-induced disruption of TJ organization of occludin and ZO-1 in differentiated Caco-2 cell monolayers was dramatically protected by epithelial growth factor (EGF) treatment through ERK activation [39]. Moreover, phosphorylated ERK by EGF is co-localized predominantly at the intercellular junctions with occludin [39], although ERK activation provides protective effects on TJ in differentiated, but not in under-differentiated Caco-2 cell monolayers [40]. In agreement with these previous studies, BJ-1108 treatment in the present study increased phosphorylation of ERK in differentiated Caco-2 cells, suggesting that BJ-1108 enhances TJ integrity via ERK activation.
The PI3K/AKT signaling pathway has been demonstrated to regulate cellular ROS and TJ proteins in IECs [41,42,43,44,45], which corroborates our findings that BJ-1108 treatment prevented phosphorylation of PI3K/mTOR, ROS generation, and TJ disruption in Caco-2 cells under inflammatory conditions. This finding was comparable with the previous observation that the treatment with BJ-1108 significantly reduced PI3K/AKT phosphorylation, ROS generation, and angiogenesis in 5-HT-treated HUVECs comparable to the reduction level by thePI3K/AKT inhibitor wortmannin [9]. Furthermore, mucosal biopsy tissue samples taken from patients with IBD show significantly higher levels of p-AKT and treatment with wortmannin alleviates disease scores and histological scores in DSS-induced colitis [46]. Given those, anticolitic effects of BJ-1108 can be possibly mediated by ROS reduction through PI3K pathway in IECs.
We presented the antioxidant effects of BJ-1108 on IECs in vitro. Given that ROS signaling is critical in the activation of immune cells such as neutrophils, macrophages, dendritic cells, T cells, and B cells [47], we cannot rule out the possibility that BJ-1108 also neutralizes harmful ROS in pro-inflammatory cells. A previous study showed that inhibition of NADPH oxidase type 2 which generates large amounts of ROS robustly reduced neutrophil infiltration and ameliorated complete Freund’s adjuvant-induced arthritis in mice [48]. In our study, we noticed that BJ-1108 reduced the infiltration of neutrophils in DSS-treated mice, as indicated by reduced MPO activity.
In conclusion, our study documented the therapeutic effects of BJ-1108 on a chemically induced experimental model of IBD. These effects may be related to the regulation of ROS stress in intestinal epithelial cells. Moreover, BJ-1108 intensified intestinal barrier function by modulating TJ molecule expression. Additionally, we found that the therapeutic action of BJ-1108 may involve the NF-kB, ERK and PI3K signaling pathway. This result supports ROS and an altered intestinal barrier as mechanisms of intestinal inflammation in part and highlights the therapeutic potential of BJ-1108 in IBD.
References
Sartor RB. Mechanisms of disease: pathogenesis of Crohn’s disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol. 2006;3:390–407.
Tian T, Wang Z, Zhang J. Pathomechanisms of oxidative stress in inflammatory bowel disease and potential antioxidant therapies. Oxid Med Cell Longev. 2017;2017:4535194.
Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol Rev. 2014;94:329–354.
Schwerd T, Bryant RV, Pandey S, et al. NOX1 loss-of-function genetic variants in patients with inflammatory bowel disease. Mucosal Immunol. 2018;11:562–574.
Kruidenier L, Kuiper I, van Duijn W, et al. Differential mucosal expression of three superoxide dismutase isoforms in inflammatory bowel disease. J Pathol. 2003;201:7–16.
Sido B, Hack V, Hochlehnert A, Lipps H, Herfarth C, Dröge W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut. 1998;42:485–492.
Serwa R, Nam TG, Valgimigli L, et al. Preparation and investigation of vitamin B6-derived aminopyridinol antioxidants. Chemistry. 2010;16:14106–14114.
Omata Y, Saito Y, Yoshida Y, et al. Action of 6-amino-3-pyridinols as novel antioxidants against free radicals and oxidative stress in solution, plasma, and cultured cells. Free Radic Biol Med. 2010;48:1358–1365.
Banskota S, Gautam J, Regmi SC, et al. BJ-1108, a 6-amino-2,4,5-trimethylpyridin-3-ol analog, inhibits serotonin-induced angiogenesis and tumor growth through PI3K/NOX pathway. PLoS ONE. 2016;11:e0148133.
Kang Y, Timilshina M, Nam TG, Jeong BS, Chang JH. BJ-1108, a 6-amino-2,4,5-trimethylpyridin-3-ol analogue, regulates differentiation of Th1 and Th17 cells to ameliorate experimental autoimmune encephalomyelitis. Biol Res. 2017;50:8.
Bhattacharyya S, Dudeja PK, Tobacman JK. ROS, Hsp27, and IKKbeta mediate dextran sodium sulfate (DSS) activation of IkappaBa, NFkappaB, and IL-8. Inflamm Bowel Dis. 2009;15:673–683.
Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Investig. 1993;69:238–249.
Graff G, Gamache DA, Brady MT, et al. Improved myeloperoxidase assay for quantitation of neutrophil influx in a rat model of endotoxin-induced uveitis. J Pharmacol Toxicol Methods. 1998;39:169–178.
Cha H, Lee S, Hwan Kim S, et al. Increased susceptibility of IDH2-deficient mice to dextran sodium sulfate-induced colitis. Redox Biol. 2017;13:32–38.
Araki Y, Sugihara H, Hattori T. In vitro effects of dextran sulfate sodium on a Caco-2 cell line and plausible mechanisms for dextran sulfate sodium-induced colitis. Oncol Rep. 2006;16:1357–1362.
Yan Y, Kolachala V, Dalmasso G, et al. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS ONE. 2009;4:e6073.
Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50:103.
Han SW, Kim JM, Lho Y, et al. DICAM attenuates experimental colitis via stabilizing junctional complex in mucosal barrier. Inflamm Bowel Dis. 2019;25:853–861.
Obermeier F, Kojouharoff G, Hans W, Schölmerich J, Gross V, Falk W. Interferon-gamma (IFN-gamma)- and tumour necrosis factor (TNF)-induced nitric oxide as toxic effector molecule in chronic dextran sulphate sodium (DSS)-induced colitis in mice. Clin Exp Immunol. 1999;116:238–245.
Wang F, Schwarz BT, Graham WV, et al. IFN-gamma-induced TNFR2 expression is required for TNF-dependent intestinal epithelial barrier dysfunction. Gastroenterology. 2006;131:1153–1163.
Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–419.
Van De Walle J, Hendrickx A, Romier B, Larondelle Y, Schneider YJ. Inflammatory parameters in Caco-2 cells: effect of stimuli nature, concentration, combination and cell differentiation. Toxicol In Vitro. 2010;24:1441–1449.
Cao M, Wang P, Sun C, He W, Wang F. Amelioration of IFN-γ and TNF-α-induced intestinal epithelial barrier dysfunction by berberine via suppression of MLCK-MLC phosphorylation signaling pathway. PLoS ONE. 2013;8:e61944.
Siddiqui A, Ancha H, Tedesco D, Lightfoot S, Stewart CA, Harty RF. Antioxidant therapy with N-acetylcysteine plus mesalamine accelerates mucosal healing in a rodent model of colitis. Dig Dis Sci. 2006;51:698–705.
Ancha HR, Kurella RR, McKimmey CC, Lightfoot S, Harty RF. Effects of N-acetylcysteine plus mesalamine on prostaglandin synthesis and nitric oxide generation in TNBS-induced colitis in rats. Dig Dis Sci. 2009;54:758–766.
You Y, Fu JJ, Meng J, Huang GD, Liu YH. Effect of N-acetylcysteine on the murine model of colitis induced by dextran sodium sulfate through up-regulating PON1 activity. Dig Dis Sci. 2009;54:1643–1650.
Amrouche-Mekkioui I, Djerdjouri B. N-acetylcysteine improves redox status, mitochondrial dysfunction, mucin-depleted crypts and epithelial hyperplasia in dextran sulfate sodium-induced oxidative colitis in mice. Eur J Pharmacol. 2012;691:209–217.
Jeong HW, Hsu KC, Lee JW, et al. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am J Physiol Endocrinol Metab. 2009;296:E955–E964.
Zhang LC, Wang Y, Tong LC, et al. Berberine alleviates dextran sodium sulfate-induced colitis by improving intestinal barrier function and reducing inflammation and oxidative stress. Exp Ther Med. 2017;13:3374–3382.
Li N, Ragheb K, Lawler G, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–8525.
Carrasco-Pozo C, Morales P, Gotteland M. Polyphenols protect the epithelial barrier function of Caco-2 cells exposed to indomethacin through the modulation of occludin and zonula occludens-1 expression. J Agric Food Chem. 2013;61:5291–5297.
Rogler G, Brand K, Vogl D, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998;115:357–369.
Neurath MF, Pettersson S, Meyer zum Büschenfelde KH, Strober W. Local administration of antisense phosphorothioate oligonucleotides to the p65 subunit of NF-kappa B abrogates established experimental colitis in mice. Nat Med. 1996;2:998–1004.
Eckmann L, Nebelsiek T, Fingerle AA, et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc Natl Acad Sci USA. 2008;105:15058–15063.
Arab HH, Al-Shorbagy MY, Abdallah DM, Nassar NN. Telmisartan attenuates colon inflammation, oxidative perturbations and apoptosis in a rat model of experimental inflammatory bowel disease. PLoS ONE. 2014;9:e97193.
Echizen K, Horiuchi K, Aoki Y, et al. NF-κB-induced NOX1 activation promotes gastric tumorigenesis through the expansion of SOX2-positive epithelial cells. Oncogene. 2019;38:4250–4263.
Lemieux E, Boucher MJ, Mongrain S, Boudreau F, Asselin C, Rivard N. Constitutive activation of the MEK/ERK pathway inhibits intestinal epithelial cell differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G719–G730.
Petecchia L, Sabatini F, Usai C, Caci E, Varesio L, Rossi GA. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab Investig. 2012;92:1140–1148.
Basuroy S, Seth A, Elias B, Naren AP, Rao R. MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J. 2006;393:69–77.
Aggarwal S, Suzuki T, Taylor WL, Bhargava A, Rao RK. Contrasting effects of ERK on tight junction integrity in differentiated and under-differentiated Caco-2 cell monolayers. Biochem J. 2011;433:51–63.
He S, Guo Y, Zhao J, et al. Ferulic acid protects against heat stress-induced intestinal epithelial barrier dysfunction in IEC-6 cells via the PI3K/Akt-mediated Nrf2/HO-1 signaling pathway. Int J Hyperthemia. 2019;35:112–121.
Park HS, Lee SH, Park D, et al. Sequential activation of phosphatidylinositol 3-kinase, beta Pix, Rac1, and Nox1 in growth factor-induced production of H2O2. Mol Cell Biol. 2004;24:4384–4394.
Zhang J, Jiang H, Xie L, et al. Antitumor effect of manumycin on colorectal cancer cells by increasing the reactive oxygen species production and blocking PI3K-AKT pathway. Onco Targets Ther. 2016;9:2885–2895.
Xu L, He S, Yin P, et al. Punicalagin induces Nrf2 translocation and HO-1 expression via PI3K/Akt, protecting rat intestinal epithelial cells from oxidative stress. Int J Hyperthermia. 2016;32:465–473.
de Araújo WM, Vidal FC, de Souza WF, de Freitas JC Jr, de Souza W, Morgado-Diaz JA. PI3K/Akt and GSK-3beta prevents in a differential fashion the malignant phenotype of colorectal cancer cells. J Cancer Res Clin Oncol. 2010;136:1773–1782.
Huang XL, Xu J, Zhang XH, et al. PI3K/Akt signaling pathway is involved in the pathogenesis of ulcerative colitis. Inflamm Res. 2011;60:727–734.
Aviello G, Knaus UG. ROS in gastrointestinal inflammation: rescue or sabotage? Br J Pharmacol. 2017;174:1704–1718.
Liu FC, Yu HP, Chen PJ, et al. A novel NOX2 inhibitor attenuates human neutrophil oxidative stress and ameliorates inflammatory arthritis in mice. Redox Biol. 2019;26:101273.
Acknowledgments
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2017R1D1A1B03028512) and by a Grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare, Republic of Korea (Grant Number: HI15C0542).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors have completed the disclosure form and declare that there are no other relationships or activities that could appear to have influenced the submitted work.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.

10620_2020_6267_MOESM1_ESM.jpg
Supplementary Figure 1.Gross image of a spleen (JPEG 553 kb)

10620_2020_6267_MOESM2_ESM.jpg
Supplementary Figure 2. LDH release by Caco-2 cells treated with BJ-1108 (0.1, 0.3, 1, 3, or 10 μM) for 48 hours (JPEG 380 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Lee, H., Lee, J.S., Cho, H.J. et al. Antioxidant Analogue 6-Amino-2,4,5-Trimethylpyridin-3-ol Ameliorates Experimental Colitis in Mice. Dig Dis Sci 66, 1022–1033 (2021). https://doi.org/10.1007/s10620-020-06267-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-020-06267-6