
Abstract
Background
The transformation of hepatic stellate cells (HSCs) into collagen-producing myofibroblasts is a key event in hepatic fibrogenesis. Recent studies have shown that microRNAs (miRNAs) play a critical role in the transformation of HSCs. However, the function of miR-489-3p in liver fibrosis remains unclear.
Methods
Here, we detected the levels of miR-489-3p and jagged canonical Notch ligand 1 (JAG1) in liver fibrosis by using CCl4-treated rats as an in vivo model and transforming growth factor-beta 1 (TGF-β1)-treated HSC cell lines LX-2 and HSC-T6 as in vitro models. The expression of profibrotic markers was affected by transfecting LX-2 cells with either miR-489-3p mimic or si-JAG1. A dual-luciferase reporter assay was carried out to study the interaction of JAG1 with miR-489-3p.
Results
We found that miR-489-3p was remarkably decreased while JAG1 was increased in liver fibrosis models both in vivo and in vitro. Overexpression of miR-489-3p reduced the expression of profibrotic markers and the activation of LX-2 cells induced by TGF-β1. Moreover, miR-489-3p decreased the expression of jagged canonical Notch ligand 1 (JAG1) in LX-2 cells by interacting with its 3ʹ-UTR. As JAG1 is a Notch ligand, decreased JAG1 by miR-489-3p inhibited the Notch signaling pathway. Moreover, the downregulation of JAG1 inhibited the expression of fibrotic markers.
Conclusion
Our results indicate that miR-489-3p can inhibit HSC activation by inhibiting the JAG1/Notch3 signaling pathway.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatic fibrosis is a programmed response to chronic liver injury caused by a viral infection, alcoholism, fatty liver disease, and autoimmune hepatitis [1, 2]. During chronic liver injury, quiescent HSCs can be activated by fibrogenic cytokines and mediators to transform into myofibroblast-like cells [3, 4]. Activated HSCs synthesize and release excessive amounts of ECM, which leads to the destruction of the normal structure of the liver and eventually develops into liver fibrosis, liver cirrhosis, and even liver cancer [5, 6]. Therefore, the exploration of potential factors affecting the activation of HSCs is useful to elucidate the underlying molecular mechanism and to develop novel antifibrogenic therapies for liver fibrosis.
The Notch signaling pathway mediates multiple different functions as an intercellular communication mediator [7]. Emerging data suggest that the Notch signaling pathway also plays essential roles in fibrotic diseases by regulating myofibroblast differentiation and epithelial/mesenchymal transition (EMT) [8, 9]. Notch signaling was activated in a liver fibrotic model and patients with fibrosis, whereas Notch inhibition significantly impeded fibrogenesis [10,11,12]. As one of the Notch ligands, JAG1 has been reported to be an inducer of fibrosis in several diseases [13,14,15] and to be elevated in cultured activated HSCs [16]. However, the role of JAG1 in liver fibrosis remains poorly defined.
MiRNAs are short (20–22 nt), noncoding, nucleotide RNA molecules [17]. Due to their pivotal effects on multiple genes and pathways, miRNAs have essential roles in diverse biological processes, including fibrosis [18, 19]. For example, miR-455 inhibits HSC activation by downregulating HSF1 expression, which is associated with the Hsp47/TGF-β/Smad4 signaling pathway [20]. MiR-489-3p is of interest to us as it has been reported to target JAG1 [21] and is upregulated in silica-induced pulmonary fibrosis and cardiac hypertrophy [22, 23]. However, no research has focused on the functions of miR-489-3p in the activation of HSCs. Overall, we hypothesized that miR-489-3p modulates HSC activation by regulating JAG1.
This study was conducted to investigate the involvement of miR-489-3p and JAG1 in HSC activation. Indeed, our findings revealed that miR-489-3p could inhibit HSC activation by inhibiting the JAG1/Notch3 signaling pathway.
Materials and Methods
Animals
Male Sprague–Dawley rats from the Experimental Center of the Hubei Medical Scientific Academy (Wuhan, China) were used to establish the carbon tetrachloride (CCl4) liver fibrosis model. Rats (180–220 g) were divided into five groups. Liver fibrosis groups were treated with a mixture of CCl4 and olive oil (1:1 vol/vol, 1 mL/kg by intraperitoneal injection twice weekly) for 2, 4, 6, 8 weeks, respectively (n = 6 in each group). The control group was intraperitoneally injected with olive oil (1 mL/kg) at the same time (n = 6). The rats were killed 3 days after the last injection to obtain liver samples.
Liver Histopathological Evaluation
Hematoxylin and eosin (H&E) staining and Masson’s trichrome staining were performed to assess liver fibrosis. Briefly, paraffin-embedded liver tissues were cut into 4 mm sections and stained with H&E staining and Masson’s trichrome staining. Liver fibrosis stages (F1–F4) were evaluated by experienced investigators.
Cell Cultures
The human HSC LX-2 cell line was obtained from the Cell Center of Shanghai Institutes for Biological Sciences. Rat HSC-T6 cells were purchased from the China Center for Type Culture Collection (Wuhan, China). HSC-T6 and LX-2 cells were cultured in DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. HSC-T6 and LX-2 cells were treated with transforming growth factor β1 (TGF-β1) (0, 2, 5, 10 ng/mL) for 24 h in DMEM with 10% FBS, respectively.
Western Blotting
The cells were collected and lysed in RIPA buffer with protease inhibitor and phosphatase inhibitor cocktail. Protein concentration was determined by using a BCA protein assay kit (Beyotime Institute of Biotechnology, Jiangsu, China). Total protein (30 mg) was mixed with sample buffer and boiled for 5 min, followed by electrophoresis with 8–12% SDS–PAGE gels and transfer to a PVDF membrane. The membrane was blocked with 5% nonfat milk for 2 h at room temperature, and blots were incubated overnight at 4 °C with primary antibodies against GAPDH, α-SMA (1:2000, Sanying Biotechnology, Wuhan, China), collagen I, Smad3, pSmad3 (1:1000, Cell Signaling Technology, Danvers, MA, USA), JAG1, Notch1, Notch2, Notch3, and Hes1 (1:1000, Abcam, Cambridge, MA, UK). After that, the membranes were incubated with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibodies (1:4000, Sanying Biotechnology, China) for 2 h at room temperature. The protein bands were visualized by using the Bio-Rad Fluro-S MultiImager.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA from liver tissues and cells was extracted using TRIzol reagent (Thermo Fisher Scientific, Santa Clara, USA). Then, the PrimeScript RT Reagent Kit (Toyobo, Osaka, Japan) was used to synthesize cDNA. Gene expression was estimated via SYBR Green real-time PCR Master Mix (Toyobo, Osaka, Japan). The primers for GAPDH were purchased from Sangon Biotech (Sangon, Shanghai, China). The specific primers (Qingke, Wuhan, China) used for qRT-PCR were as follows: human: α-SMA, forward: 5ʹ-CTCTGGACGCACAACTGGCATC-3ʹ, reverse: 5ʹ-CACGCTCAGCAGTAGTAACGAAGG-3ʹ; collagen I, forward: 5ʹ-GCGAGAGCATGACCGATGGATTC-3ʹ, reverse: 5ʹ-GCCTTCTTGAGGTTGCCAGTCTG-3ʹ; JAG1, forward: 5ʹ-TAAACGCCAAATCCTGTAAGA-3ʹ, reverse: 5ʹ-CAGCGATAACCATTAACCAAA-3ʹ; Notch3, forward: 5ʹ-ACCTGCCTCAACACACCT-3ʹ, reverse: 5ʹ-ATTCTGACCCTCAAACCC-3ʹ; and Hes1, forward: 5ʹ-GGCTGGAGAGGCGGCTAAGG-3ʹ, reverse: 5ʹ-TGCTGGTGTAGACGGGGATGAC-3ʹ. Rat: jagged1, forward: 5ʹ-ATGCCTCCTGTCGGGATTTG-3ʹ, reverse: 5ʹ- AGTGACCCCCATTCAAGCAG-3ʹ. The primers for U6 small nucleolar RNA and miR-489-3p were purchased from RiboBio (Guangzhou, China) and used as a control. The expression of miRNAs and mRNAs was determined using the 2−ΔΔCt method.
siRNA and miRNA Mimics Transfection
Specific small interfering RNA (siRNA) targeting JAG1, miR-489-3p mimics, and their controls were synthesized by RiboBio (Guangzhou, China). Cells were transfected with siRNA or miRNA mimic using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. After 24 h, the cells were treated with drugs.
Luciferase Activity Assay
The experiments were repeated independently three or more times. The data are represented as the mean and standard deviation (SD). GraphPad Prism 5 software (GraphPad, USA) was used to perform the statistical analyses. P < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Statistical Analysis
Independent experiments were repeated three or more times. The data are represented as the mean and standard deviation (SD). GraphPad Prism 5 software (GraphPad, USA) was used to assess statistical analyses. P < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
MiR-489-3p Was Downregulated and JAG1 Was Upregulated in the Progression of Liver Fibrogenesis in Rats
To establish a liver fibrosis model, male Sprague–Dawley rats were treated with CCl4. Hematoxylin and eosin staining (H&E) and Masson’s trichrome staining were used to assess the degree of liver fibrosis. Typical liver fibrosis stages (F0–F4) are illustrated in Fig. 1a. The qRT-PCR results showed that miR-489-3p was significantly downregulated in liver fibrotic tissues compared with the control group (Fig. 1b). Conversely, JAG1 expression was increased in liver fibrosis induced by CCl4 (Fig. 1c). These results indicate that miR-489-3p and JAG1 might participate in the process of liver fibrosis.

MiR-489-3p was downregulated and JAG1 was upregulated in the progression of liver fibrogenesis in rats. Rats were injected intraperitoneally with CCl4. a Representative H&E and Masson’s trichrome-stained images at ×40 and ×100 magnification. qRT-PCR measured miR-489-3p (B) and JAG1 c expression in liver fibrosis stages F0 (n = 6), F1–3 (n = 12), and F4 (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001
MiR-489-3p and JAG1 Were Dysregulated in TGF-β1-Activated HSCs
LX-2 and HSC-T6 cells were treated with TGF-β1 (0, 2, 5, 10 ng/mL) for 24 h. We found that both the mRNA (Fig. 2a, b, d, e) and the protein (Fig. 2h) levels of α-SMA and collagen I were significantly increased compared to control cells. These results confirmed that LX-2 and HSC-T6 cells were successfully activated by TGF-β1. MiR-489-3p expression showed a dose-dependent decrease in activated LX-2 (Fig. 2c) and HSC-T6 (Fig. 2f) cells. The mRNA (Fig. 2g) and protein (Fig. 2h) expression of JAG1 also remarkably increased when TGF-β1 was used to further stimulate LX-2 cell activation. These results demonstrate that miR-489-3p is downregulated and JAG1 is upregulated in TGF-β1-activated HSCs.

MiR-489-3p and JAG1 are dysregulated in TGF-β1-activated HSCs. LX-2 and HSC-T6 cells were treated with 0, 2, 5, or 10 ng/ml TGF-β1 for 24 h. The mRNA levels of α-SMA (a, d), collagen I (b, e), and JAG1 (g) were analyzed by qRT-PCR. Relative miR-489-3p expression in activated HSC-T6 (c) and LX-2 (F) cells were examined in a dose-dependent manner. α-SMA, collagen I, and JAG1 protein (H) levels were determined by western blotting. *P < 0.05, **P < 0.01, ***P < 0.001
MiR-489-3p Inhibits the Activation of LX-2 Cells Induced by TGF-β1
To identify the role of miR-489-3p in HSC activation, LX-2 cells were transfected with miR-489-3p mimics or miR-negative control (NC) (50 μM). As expected, miR-489-3p expression was markedly upregulated by the mimics compared with the control (Fig. 3a). The mRNA expression levels of α-SMA, collagen I, and Smad3 were decreased following the overexpression of miR-489-3p in LX-2 cells (Fig. 3b). The protein levels of pSmad3, α-SMA, and collagen I were also reduced (Fig. 3c). However, the protein level of Smad3 remained the same. To determine whether miR-489-3p can also influence TGF-β1-induced HSC activation, LX-2 cells were transfected with miR-489-3p mimics for 24 h and then treated with 5 ng/ml TGF-β1 for 24 h. Strikingly, overexpression of miR-489-3p exhibited a significant downregulation of both α-SMA and collagen I (Fig. 3d). These results demonstrate that miR-489-3p can suppress the expression of profibrotic markers and the activation of LX-2 cells induced by TGF-β1.

MiR-489-3p inhibits the activation of LX-2 cells induced by TGF-β1. LX2 cells were transfected with miR-489-3p mimic or miR-negative control for 24 or 48 h. The expression level of miR-489-3p (a) was examined by qRT-PCR. The mRNA (b) expression of the profibrotic markers α-SMA, collagen I, and Smad3 was analyzed by qRT-PCR. The protein (c) levels of α-SMA, collagen I, Smad3, and pSmad3 were determined by western blotting. LX2 cells were transiently transfected with miR-489-3p for 24 h and then treated with 5 ng/ml TGF-β1 for 24 h. The protein (d) level of profibrotic markers was determined by western blotting. *P < 0.05, **P < 0.01, ***P < 0.001
MiR-489-3p Directly Targets JAG1 and Suppresses JAG1/Notch3 Signaling
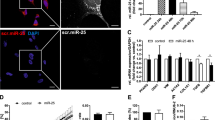
Previous studies have reported that miR-489-3p directly targets JAG1 in bladder cancer cells [21]. To investigate whether miR-489-3p could bind to the 3′ untranslated region (3′UTR) of JAG1 in LX-2 cells, we cloned the 3′UTR sequences of human JAG1 mRNA into the pmirGLO vector (Fig. 4a). Luciferase activity was reduced by cotransfection of the wild-type plasmids and miR-489-3p mimic (Fig. 4b). Conversely, the mutant plasmids showed no significant change in luciferase activity in LX-2 cells (Fig. 4b). The expression of JAG1 at both the RNA (Fig. 4c) and protein (Fig. 4d) levels was markedly decreased in miR-489-3p mimic-transfected LX-2 cells. This implies that miR-489-3p regulates the expression of JAG1 at the transcriptional and posttranscriptional levels. In addition, Notch3 and the downstream target gene Hes1 were also reduced in miR-489-3p mimic-transfected cells, while the expression of Notch1 and Notch2 was unchanged (Fig. 4c, d). These results indicate that miR-489-3p directly binds to JAG1 mRNA and suppresses JAG1/Notch3 signaling.

MiR-489-3p directly targets JAG1 and suppresses JAG1/Notch3 signaling. Predicted miR-489-3p targeting sequence in the JAG1 (WT or mutant) 3′-UTR (a). Luciferase reporter assay of LX2 cells transfected with pmirGLO vectors carrying JAG1 WT or mutant binding site together with miR-489-3p mimics or NC mimic (n = 3) (b). Relative expression levels of JAG1, Notch3, and Hes1 were examined in LX-2 cells by qRT-PCR (c). The protein levels of JAG1, Notch1, Notch2, Notch3, and Hes1 were determined by western blotting (d). *P < 0.05, **P < 0.01, ***P < 0.001
Inhibition of JAG1 Blockade of Fibroblast Activation
JAG1 expression increased significantly in a rat liver fibrosis model and TGF-β1-activated LX-2 cells. To elucidate the biological function of JAG1 in the activation of HSCs, we constructed a small interfering RNA (siRNA) to reduce JAG1 mRNA (Fig. 5a) and protein (Fig. 5b) expression and transfected it into LX-2 cells. As illustrated in Fig. 5c, d, inhibition of JAG1 reduced the expression of fibrotic markers. These data demonstrate that JAG1 is necessary for HSC activation.

Inhibition of JAG1 blockade of fibroblast activation. LX2 cells were transfected with si-JAG1 or si-NC for 24 and 48 h. JAG1 expression was examined by qRT-PCR (a) and western blotting (b). Relative expression levels of profibrotic markers were analyzed by qRT-PCR (b). The protein (c) level of profibrotic markers was determined by western blotting. *P < 0.05, **P < 0.01, ***P < 0.001
Discussion
Although advanced liver fibrosis was historically thought to be an irreversible process, increasing evidence suggests that cirrhosis can be reversed if the underlying causes of liver damage are addressed [24]. With HSCs positioned at the nexus of hepatic fibrosis, a comprehensive understanding of their complex biology is therefore crucial to the design of novel antifibrotic therapies.
MiR-489-3p was found to be a regulator of cancer progression in a number of tumors. Several reports have shown that miR-489-3p suppresses fibrosis of the lung and heart [22, 23]. However, no research has focused on the functions of miR-489-3p in liver fibrosis. Our results showed that miR-489-3p was downregulated in liver fibrosis models both in vivo and in vitro. Overexpression of miR-489-3p inhibited the activation of HSCs. These results indicate that miR-489-3p participates in the activation of HSCs.
To date, Smad3 and MyD88 have been identified as the target genes of miR-489-3p in fibrosis [22, 23]. However, we found that miR-489-3p did not change the expression of Smad3 in LX-2 cells. This may be because miR-489-3p has different targets in lung and liver tissues since one miRNA can have multiple targets. JAG1 was reported to be a target of miR-489-3p in HEK293 cells [21]. Our data also showed that miR-489-3p directly targeted JAG1 in LX-2 cells. Interestingly, the Notch receptor, Notch3, and its downstream target gene Hes1 were also reduced in the miR-489-3p mimic-transfected LX-2 cells. This implies that miR-489-3p might inhibit the activation of HSCs by directly targeting JAG1 and suppressing JAG1/Notch3 signaling.
The Notch signaling pathway is an evolutionarily highly conserved system [25]. Recent reports have implied that the expression of JAG1 is elevated in cultured HSCs that develop into myofibroblast-like cells [16]. And Huang [26] found that JAG1 and Notch2 were closely related to the degree of renal interstitial fibrosis, and the deletion of JAG1 or Notch2 protected folic acid-induced kidney fibrosis in mice. Our studies found that JAG1 was upregulated in the CCl4-treated rat model, as well as in TGF-β1-induced activated HSCs. We showed that JAG1 siRNA transfection decreased the expression of profibrotic genes in LX-2 cells. Considering that each Notch molecule produces one signal through proteolysis and so can only signal once, restriction of ligand and/or receptor expression is a simple way of controlling Notch activation [27]. Inhibition of Notch activation by JAG1 downregulation might restrict HSC activation by reducing the transcription of genes associated with fibrosis. The activation of Notch signaling induces transcriptional activation of the Notch target gene Hes1. Overexpression of Hes1 stimulated the promoter activities of profibrotic genes in HSC-T6 cells [28]. In addition to this direct effect mediated by its intracellular domain, Notch can indirectly regulate the development of liver fibrosis through cross talk with TGF-β1 [29]. The TGF-β1 signaling pathway is a classical pathway for the activation of HSCs [30]. We demonstrated that miR-489-3p could inhibit HSC activation by affecting JAG1 expression and JAG1/Notch3 signaling. Further studies are needed to evaluate the therapeutic potential of miR-489-3p in animal models.
In summary, we found that restoration of miR-489-3p could reduce HSC activation by regulating JAG1 expression and suppressing JAG1/Notch3 signaling (Fig. 6). To further demonstrate the importance of this novel mechanism, future studies are needed to evaluate the effect of miR489-3p/JAG1 signaling in hepatic fibrosis models in vivo.

Schematic. The Notch signaling pathway is activated when JAG1 interacts with Notch3 and releases the Notch intracellular domain (NICD) to activate transcription of the Notch target gene helix–loop–helix transcription factor Hes1. In addition to this direct effect, Notch can indirectly regulate the activation of HSCs through cross talk with the TGF-β/Smad3 signaling pathway. MiR-489-3p inhibits the activation of HSCs by directly targeting JAG1 and suppressing JAG1/Notch3 signaling
References
Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet (London, England). 2014;383:1749–1761. https://doi.org/10.1016/s0140-6736(14)60121-5.
Pimpin L, Cortez-Pinto H, Negro F, et al. Burden of liver disease in Europe: epidemiology and analysis of risk factors to identify prevention policies. J Hepatol. 2018;69:718–735. https://doi.org/10.1016/j.jhep.2018.05.011.
Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. https://doi.org/10.1038/nrgastro.2017.38.
Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3:1473–1492. https://doi.org/10.1002/cphy.c120035.
Tsukada S, Parsons CJ, Rippe RA. Mechanisms of liver fibrosis. Clinica chimica acta. Int J Clin Chem. 2006;364:33–60. https://doi.org/10.1016/j.cca.2005.06.014.
Pinheiro D, Dias I, Ribeiro Silva K, et al. Mechanisms underlying cell therapy in liver fibrosis: an overview. Cells.. 2019;. https://doi.org/10.3390/cells8111339.
Meurette O, Mehlen P. Notch Signaling in the tumor microenvironment. Cancer Cell.. 2018;34:536–548. https://doi.org/10.1016/j.ccell.2018.07.009.
Hu B, Phan SH. Notch in fibrosis and as a target of anti-fibrotic therapy. Pharmacol Res. 2016;108:57–64. https://doi.org/10.1016/j.phrs.2016.04.010.
Edeling M, Ragi G, Huang S, Pavenstadt H, Susztak K. Developmental signalling pathways in renal fibrosis: the roles of Notch, Wnt and Hedgehog. Nature Reviews Nephrology.. 2016;12:426–439. https://doi.org/10.1038/nrneph.2016.54.
Chen Y, Zheng S, Qi D, et al. Inhibition of Notch signaling by a gamma-secretase inhibitor attenuates hepatic fibrosis in rats. PloS ONE. 2012;7:e46512. https://doi.org/10.1371/journal.pone.0046512.
Chen YX, Weng ZH, Zhang SL. Notch3 regulates the activation of hepatic stellate cells. World J Gastroenterol. 2012;18:1397–1403. https://doi.org/10.3748/wjg.v18.i12.1397.
Huang M, Chang A, Choi M, Zhou D, Anania FA, Shin CH. Antagonistic interaction between Wnt and Notch activity modulates the regenerative capacity of a zebrafish fibrotic liver model. Hepatology (Baltimore, Md).. 2014;60:1753–1766. https://doi.org/10.1002/hep.27285.
Condorelli AG, Logli E, Cianfarani F, et al. MicroRNA-145-5p regulates fibrotic features of recessive dystrophic epidermolysis bullosa skin fibroblasts. Br J Dermatol. 2019;181:1017–1027. https://doi.org/10.1111/bjd.17840.
Chen X, Xiao W, Chen W, et al. MicroRNA-26a and -26b inhibit lens fibrosis and cataract by negatively regulating Jagged-1/Notch signaling pathway. Cell Death Differ. 2017;24:1431–1442. https://doi.org/10.1038/cdd.2016.152.
Zhao S, Xiao X, Sun S, et al. MicroRNA-30d/JAG1 axis modulates pulmonary fibrosis through Notch signaling pathway. Pathol Res Pract. 2018;214:1315–1323. https://doi.org/10.1016/j.prp.2018.02.014.
Sawitza I, Kordes C, Reister S, Haussinger D. The niche of stellate cells within rat liver. Hepatology (Baltimore, Md).. 2009;50:1617–1624. https://doi.org/10.1002/hep.23184.
Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol cell Biol. 2014;15:509–524. https://doi.org/10.1038/nrm3838.
Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol cell Biol. 2019;20:21–37. https://doi.org/10.1038/s41580-018-0045-7.
Piperigkou Z, Gotte M, Theocharis AD, Karamanos NK. Insights into the key roles of epigenetics in matrix macromolecules-associated wound healing. Adv Drug Deliv Rev. 2018;129:16–36. https://doi.org/10.1016/j.addr.2017.10.008.
Wei S, Wang Q, Zhou H, et al. miR-455-3p alleviates hepatic stellate cell activation and liver fibrosis by suppressing HSF1 expression. Mol Ther Nucleic Acids. 2019;16:758–769. https://doi.org/10.1016/j.omtn.2019.05.001.
Li J, Qu W, Jiang Y, et al. miR-489 Suppresses proliferation and invasion of human bladder cancer cells. Oncol Res. 2016;24:391–398. https://doi.org/10.3727/096504016x14666990347518.
Wu Q, Han L, Yan W, et al. miR-489 inhibits silica-induced pulmonary fibrosis by targeting MyD88 and Smad3 and is negatively regulated by lncRNA CHRF. Sci Rep. 2016;6:30921. https://doi.org/10.1038/srep30921.
Wang K, Liu F, Zhou LY, et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377–1388. https://doi.org/10.1161/circresaha.114.302476.
Marcellin P, Gane E, Buti M, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet (London, England).. 2013;381:468–751. https://doi.org/10.1016/s0140-6736(12)61425-1.
Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012;13:654–666. https://doi.org/10.1038/nrg3272.
Huang S, Park J, Qiu C, et al. Jagged1/Notch2 controls kidney fibrosis via Tfam-mediated metabolic reprogramming. PLoS Biol. 2018;16:e2005233. https://doi.org/10.1371/journal.pbio.2005233.
Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. https://doi.org/10.1016/j.cell.2009.03.045.
Zhang K, Zhang YQ, Ai WB, et al. Hes1, an important gene for activation of hepatic stellate cells, is regulated by Notch1 and TGF-beta/BMP signaling. World J Gastroenterol. 2015;21:878–887. https://doi.org/10.3748/wjg.v21.i3.878.
Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. https://doi.org/10.1038/cr.2008.302.
Dewidar B, Meyer C, Dooley S, Meindl-Beinker AN. TGF-beta in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells. 2019;. https://doi.org/10.3390/cells8111419.
Funding
This work was supported by the Innovation Cultivation Program of Zhongnan Hospital of Wuhan University [znpy2018095] and the National Natural Science Foundation of China [81670554].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All the authors declare that there is no conflict of interest in this article.
Ethical approval
The Committee on the Ethics of Animal Experiments of the Wuhan University School of Medicine approved the animal experimental procedures (permit number: 2017055).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, J., Dong, S., Ye, M. et al. MicroRNA-489-3p Represses Hepatic Stellate Cells Activation by Negatively Regulating the JAG1/Notch3 Signaling Pathway. Dig Dis Sci 66, 143–150 (2021). https://doi.org/10.1007/s10620-020-06174-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-020-06174-w