
Abstract
Background
Allyphenyline, a novel α2-adrenoceptor (AR) ligand, has been shown to selectively activate α2C-adrenoceptors (AR) and 5HT1A receptors, but also to behave as a neutral antagonist of α2A-ARs. We exploited this unique pharmacological profile to analyze the role of α2C-ARs and 5HT1A receptors in the regulation of gastric mucosal integrity and gastrointestinal motility.
Methods
Gastric injury was induced by acidified ethanol in Wistar rats. Mucosal catalase and superoxide dismutase levels were measured by assay kits. The effect of allyphenyline on electrical field stimulation (EFS)-induced fundic and colonic contractions was determined in C57BL/6 mice.
Results
Intracerebroventricularly injected allyphenyline (3 and 15 nmol/rat) dose dependently inhibited the development of mucosal damage, which was antagonized by ARC 239 (α2B/C-AR and 5HT1A receptor antagonist), (S)-WAY 100135 (selective 5HT1A receptor antagonist), and JP-1302 (selective α2C-AR antagonist). This protection was accompanied by significant elevation of mucosal catalase and superoxide dismutase levels. Allyphenyline (10−9–10−5 M) also inhibited EFS-induced fundic contractions, which was antagonized by ARC 239 and (S)-WAY 100135, but not by JP-1302. Similar inhibition was observed in the colon; however, in this case only ARC 239 reduced this effect, while neither selective inhibition of α2C-ARs and 5HT1A receptors nor genetic deletion of α2A- and α2B-ARs influenced it.
Conclusions
Activation of both central α2C-ARs and 5HT1A receptors contributes to the gastroprotective action of allyphenyline in rats. Its inhibitory effect on fundic contractions is mediated by 5HT1A receptors, but neither α2-ARs nor 5HT1A receptors take part in its inhibitory effect on colonic contractility in mice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
α2-adrenoceptors (α2-ARs) belong to the superfamily of G-protein-coupled receptors and consist of three different molecular subtypes (α2A, α2B, and α2C) [1], which are widely distributed in the central nervous system (CNS) and in peripheral tissues and possess distinct functional and pharmacological properties (for reviews see [2–4]). The synthesis of subtype-selective ligands and generation of genetically engineered mice have allowed significant advances in our understanding of the role of each subtype. Several studies demonstrated that most of the classical effects of α2-AR agonists, such as hypotension, bradycardia, or sedation are mediated entirely or predominantly by the α2A-subtype [2, 4].
Since the pioneering work of Paton and Vizi [5], who reported the localization of presynaptic α2-ARs on cholinergic myenteric nerves and their inhibitory action on the release of acetylcholine, a vast amount of data have been accumulated on the role of these receptors in the gastrointestinal (GI) tract as well. Besides cholinergic neurons, α2-ARs are also localized on extrinsic noradrenergic neurones (autoreceptors), as well as on enterocytes and smooth muscles, and their activation results in various effects, such as inhibition of GI motility, secretion and visceral sensation, and increased intestinal absorption of water and electrolytes [6, 7]. Moreover, there is evidence that α2-ARs localized in the CNS are also involved in the inhibition of GI motility and secretion [8–10]. Due to all these actions, α2-ARs are considered attractive therapeutic targets for the treatment of a wide range of GI disorders, including functional dyspepsia, irritable bowel syndrome, and inflammatory bowel diseases [7, 11]. However, most GI effects are mediated mainly by the α2A-AR subtype [4, 7, 12], and pharmacologic modulation of this subtype in the above-mentioned diseases by non-selective or subtype-selective α2-AR ligands may result in numerous undesirable side effects outside the GI tract (see above).
In the last two decades, our research group demonstrated that α2-AR agonists induce a potent defensive effect in the stomach and reduce the extent of mucosal injury in both acid-dependent (indomethacin) and acid-independent (ethanol) ulcer models [12–17]. More importantly, our results clearly showed that this protective effect is mediated by α2B- and α2C-ARs localized in the brain, most probably in the dorsal vagal complex, and α2A-ARs do not have any significant role in it [12, 14, 17]. Accordingly, we proposed that selective α2B/C-AR agonists may represent a novel group of gastroprotective agents, which are devoid of several side effects mediated by the α2A-AR subtype, such as hypotension, bradycardia, or sedation [17]. However, although the site of gastroprotective action is likely to be central, it should be considered that α2B-ARs are located also postsynaptically on vessels [18, 19] and appear to be involved in the vasoconstrictor response to α2-AR agonists [20] and in salt-induced hypertension [21]. Consequently, selective α2C-AR stimulants are likely to be even more favorable and safer potential therapeutic agents against gastric mucosal injury than the mixed α2B/C-AR agonists, since they are devoid of adverse effects due to activation of both α2A- and α2B-ARs.
In the recent years, allyphenyline, a novel α2-AR ligand, endowed with peculiar pharmacological profile has been synthetized and characterized [22–24]. Among the three α2-AR subtypes, allyphenyline behaves as an agonist only at the α2C-AR subtype, while it does not bind to α2B-ARs and behaves as a neutral antagonist at the α2A-AR subtype in vitro [22]. In line with it, allyphenyline enhanced morphine analgesia and attenuated morphine withdrawal symptoms in vivo (due to its α2C-AR agonist property), but it was devoid of any sedative effect (due to its α2A-AR-antagonism) [23, 24].
Regarding other receptors, allyphenyline also binds to and activate 5HT1A receptors [25], and dual activation of α2C-ARs and 5HT1A receptors by this compound elicited antidepressant- and anxiolytic-like effects in rodents [25, 26]. 5HT1A receptors have been implicated in numerous GI processes, such as gastric mucosal protection [27, 28], inhibition of cholinergic gastric contractions [29, 30], as well as modulation of colonic motility [31]. It should be emphasized that ulcer disease is often accompanied by increased gastric motility [32], consequently inhibition of GI motility may contribute to the therapeutic effect of an anti-ulcer agent.
Based on the receptorial actions, we hypothesized that allyphenyline may induce gastroprotective action (due to dual activation of α2C-ARs and 5HT1A receptors) and may also inhibit GI motility, in spite of the lack of α2A-AR activation (due to 5HT1A receptor agonism).
Our results confirmed our expectations and demonstrated that (1) allyphenyline potently inhibits the development of ethanol-induced mucosal damage, and central α2C-ARs and 5HT1A receptors are both involved in this action, (2) allyphenyline concentration dependently suppresses cholinergic contractions in the stomach, which effect is—at least partly—mediated by 5HT1A receptors, but not by α2C-ARs, and (3) allyphenyline also suppresses cholinergic contractions in the colon, but this effect does not depend on the activation of 5HT1A receptors and α2-ARs. Hence, the unique receptorial profile makes allyphenyline particularly attractive for the treatment of various GI disorders, such as peptic ulcers, functional dyspepsia (FD), or irritable bowel syndrome (IBS), because both mucosal protection, fundic relaxation, and inhibition of colonic motility can be achieved without inducing α2A- and α2B-AR-mediated unwanted side effects.
Methods
Synthesis of Allyphenyline
Allyphenyline (2-(1-(2-allylphenoxy)ethyl)-4,5-dihydro-1H-imidazole) [22] was obtained from 2-(2-allylphenoxy)propanenitrile [33] by treatment with sodium methoxide in methanol and subsequent condensation with ethylenediamine, as reported in Fig. 1.

Synthesis of allyphenyline (50 % yield). The free base was transformed into the oxalate salt; which was recrystallized from 2-PrOH: mp 154–155 °C. 1H NMR (DMSO): δ 1.53 (d, 3, CH 3), 3.42 (m, 2, CH 2CH), 3.88 (s, 4, NCH 2CH 2N), 5.06 (dd, 2, CH = CH 2), 5.40 (q, 1, OCH), 5.95 (m, 1, CH = CH2), 6.92–7.26 (m, 4, ArH), 7.81 (br s, 1, NH, exchangeable with D2O). Anal. Calcd for C14H18N2O H2C2O4: C, 56.80; H, 6.55; N, 8.28. Found: C, 56.69; H, 6.71; N, 8.13
Animals
For in vivo analysis of gastric mucosal defensive processes, male Wistar rats weighing 150–170 g (Semmelweis University) were used [15], while for ex vivo analysis of gastric and colonic contractility C57BL/6 mice from both sexes (20–25 g, National Institute of Oncology) were used [34]. α2A- and α2B-AR knockout mice (with the same C57BL/6 genetic background) were the generous gift of Prof. L. Hein (University of Freiburg, Germany). The generation of these mouse lines has been described previously in detail [20, 35]. Genotypes were confirmed by subtype-specific polymerase chain reactions (with a TProfessional Basic Thermocycler, Biometra) performed with genomic DNA isolated from tail biopsies as described previously [36]. Animals were housed in a temperature- and humidity-controlled room at a 12-h light/dark cycle under conditions of animal housing and experimentation according to ethical guidelines issued by the Ethical Board of Semmelweis University, based on EC Directive 86/609/EEC.
All procedures conformed to the European convention for the protection of vertebrate animals used for experimental and other scientific purposes, and all efforts were made to minimize the suffering of animals. The experiments were approved by the National Scientific Ethical Committee on Animal Experimentation and permitted by the government (Food Chain Safety and Animal Health Directorate of the Central Agricultural Office (PEI/001/1493-4/2015).
In Vivo Analysis of Gastric Mucosal Protective Processes
Before the experiments, rats were deprived of food for 24 h with free access to tap water. Gastric mucosal lesions were induced by acidified ethanol (98 ml absolute ethanol + 2 ml concentrated HCl), which was given intragastrically in a volume of 0.5 ml/rat by an oral gavage using a stainless steel cannula. All test compounds were injected into the lateral brain ventricle (intracerebroventricularly, i.c.v.) 10 min before the ethanol challenge in a volume of 10 μl, as described previously [15]. 60 min after the injection of ethanol, the animals were killed, and the stomachs were excised and opened along the greater curvature. The flattened stomachs were photographed and pixel numbers of the damaged and total mucosal areas were determined using a free photo-editing software. The percentage of the damaged mucosal area in each stomach was calculated as follows: [(pixel number of ulcerated mucosa/pixel number of total mucosal area) × 100]. After the stomachs were photographed, gastric mucosa was separated on a cooled plate, shock-frozen in liquid nitrogen, and stored at −80 °C until further assayed.
Study Design
In four consecutive experiments, a total of 80 rats were randomly divided into 16 groups (four groups/experiment, five rats/group). In the first experiment, rats received water per os and vehicle i.c.v. (absolute control group), acidified ethanol per os and vehicle i.c.v. (ethanol control group), or acidified ethanol per os and allyphenyline i.c.v. at 2 different doses (3 and 15 nmol). In the three other experiments, rats received acidified ethanol per os and vehicle, allyphenyline (15 nmol), an α2-AR and/or 5HT1A receptor antagonist, or the combination of allyphenyline and the respective antagonist i.c.v. The applied doses of drugs were selected based partly on our preliminary results, partly on the literature data [16, 17, 25, 26].
Catalase Assay
Catalase (CAT) enzyme activity was measured by using an assay kit (Cayman Europe, Tallinn, Estonia) according to the manufacturer’s instructions. This kit utilizes the peroxidatic function of CAT for determination of enzyme activity. The method is based on the reaction of the enzyme with methanol in the presence of an optimal concentration of H2O2. The formaldehyde produced was measured spectrophotometrically with 4-amino-3-hydrazino-5-mercapto-1,2,4-triazole as the chromogen. One unit of CAT was defined as the amount of enzyme that causes the formation of 1 nmol of formaldehyde per minute at 25 °C. CAT activity was expressed in nmol/min/mg tissue.
Superoxide Dismutase Assay
Superoxide dismutase (SOD) activity was measured by using an assay kit (Cayman Europe, Tallinn, Estonia) according to the manufacturer’s instructions. This kit utilizes a tetrazolium salt for the detection of superoxide radicals generated by xanthine oxidase and hypoxanthine, and measures the activity of all three types of SOD (Cu/Zn, Mn, and Fe-SOD). One unit of SOD was defined as the amount of enzyme needed to exhibit 50 % dismutation of the superoxide radical (O ·−2 ). SOD activity was expressed in unit/mg tissue.
Ex Vivo Analysis of Fundic Contractility
The preparation of gastric fundus strips was performed as described previously [34]. Briefly, mice were killed by cervical dislocation, and the entire stomach was removed and placed in Petri dish containing Krebs solution and aerated with carbogen (mixture of 95 % O2 and 5 % CO2). The fundal part was separated from the pyloric part of each stomach, and 2- to 2.5-cm-long fundus strips were prepared by transverse cuts on each side of the strip parallel to the longitudinal muscle. From each stomach, only one strip was prepared.
The isolated gastric fundus strips were suspended between upper (ring) and lower (straight) electrodes in organ baths of 5 ml volume containing Krebs solution aerated with carbogen at 37 °C. Krebs solution has the following composition (mM/L): NaCl, 118.0; NaHCO3, 25.0; KCl, 4.7; KH2PO4, 1.2; glucose, 11.0; CaCl2, 2.5; MgSO4, 1.2. The upper end of the strip was attached by thread to a transducer, connected to a computer through an amplifier. The resting tension was adjusted to 0.5 g. To induce contraction of gastric fundus strip, electrical field stimulation (EFS) was used with the following stimulation parameters: pairs (200 ms pulse distance) of rectangular impulses (1 ms pulse width, 25 shocks, 9 V/cm, i.e., supramaximal intensity of 5 Hz) were repeated in 60 s. Each impulse induced a fast contractile response, followed by a slower relaxation response, whose amplitudes were constant over a period of 60 min (pilot experiments). The contractions were abolished by atropine (1 µM), indicating the predominant role of the cholinergic system in the contractile response, while the relaxations were inhibited by Nω-Nitro-l-arginine (L-NNA, 10 µM), indicating the activation of nitrergic pathways.
After setting up, strips were allowed to equilibrate for 30–45 min before adding the first concentration of allyphenyline or clonidine (non-selective α2-AR agonist, used as a reference compound). A period of 5 min was allowed to elapse between concentration increment. Cumulative concentration–response curves were constructed for both agonists. When the effects of the antagonists (ARC 239, (S)-WAY 100135, and JP-1302) on the inhibitory effect of allyphenyline were assessed, the antagonists (1 µM) were added to the organ bath 20 min prior the first concentration of allyphenyline. The antagonists in this concentration were effective and selective [34, 37–39]. Drugs were given in volumes <1 % of total bath volume.
The 50 % effective concentrations (EC50) and maximal inhibitory effect (E max) of agonists were calculated from the nonlinear regression of individual sigmoid concentration response curves. The average of five peaks of contractions before adding the agonists was taken as 100 % (control). The inhibitory effect of the agonists (the mean of five peaks) was expressed as a percentage to the control contractions.
Ex Vivo Analysis of Colonic Contractility
Besides the stomach, the most distal (~2 cm) segment of the colon was also removed from the same mouse. The same steps were followed as in the preceding case, but based on preliminary studies stimulation parameters were modified, and single rectangular impulses (1 ms pulse width, 1 shock, 9 V/cm, i.e., supramaximal intensity of 5 Hz) were repeated in 30 s. These impulses induced a fast, slight relaxation response followed by a contractile response, with constant amplitudes over a period of 60 min, and were abolished by L-NNA (10 µM) and atropine (1 µM), respectively.
Chemical Compounds
Clonidine hydrochloride (non-selective α2-AR agonist, PubChem CID 2803) and JP-1302 trihydrochloride hydrate (selective α2C-AR antagonist, PubChem CID 540335) were purchased from Sigma-Aldrich (St. Louis, USA), while (S)-WAY 100135 dihydrochloride (selective 5HT1A receptor antagonist, PubChem CID 6604840) and ARC 239 dihydrochloride (α2B/C-AR and 5HT1A receptor antagonist, PubChem CID 609483) were obtained from Tocris Bioscience (Bristol, UK). Allyphenyline (PubChem CID 24906198) was synthetized as described above. All drugs were dissolved in saline (in vivo) or in distilled water (ex vivo). For controls the respective vehicles were used.
Statistical Analysis
Data are expressed as mean ± SEM. Statistical analysis of the data was performed either with Student’s t test (two treatment groups), or with one-way ANOVA followed by Newman–Keuls post hoc test, or (in the case of comparing concentration–response curves) by two-way repeated measures ANOVA followed by Holm-Sidak post hoc test. A probability of p < 0.05 was considered statistically significant.
Results
The Effect of Allyphenyline on Ethanol-Induced Mucosal Damage
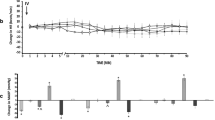
Oral administration of acidified ethanol induced multiple longitudinal hemorrhagic lesions on the gastric mucosa (damaged mucosal area: 9.3 ± 1.9 %, n = 20, Fig. 2). I.c.v. injection of allyphenyline (3 and 15 nmol/rat) 10 min before the ethanol challenge dose dependently reduced the development of mucosal lesions, and at the higher dose, it induced almost complete inhibition (damaged areas: 1.4 ± 0.5 and 0.2 ± 0.1 %, respectively, n = 5, p < 0.01, which correspond to 21.7 and 4 % of the damaged area in the respective ethanol control group, Fig. 2).

a The effect of allyphenyline (ALL, 3 and 15 nmol/rat) on ethanol (ETH)-induced gastric mucosal injury in the absence and presence of ARC 239 (ARC, 50 nmol/rat), (S)-WAY 100135 (WAY, 50 nmol/rat), and JP-1302 (JP, 56 nmol/rat). Allyphenyline was injected intracerebroventricularly (i.c.v.) either alone or together with the respective antagonists 10 min before the ethanol challenge. In order to compare the inhibitory effects of the antagonists used in three different experiments, the damaged mucosal area of each stomach was normalized to the mean value of the respective ethanol control group (100 %). Each column represents mean (±SEM) of 5 rats. **p < 0.01 compared with ethanol-treated group (column 2); # p < 0.05, ## p < 0.01 compared with allyphenyline 15 nmol-treated group (column 4) (ANOVA, Newman–Keuls post hoc test). b Representative macroscopic pictures demonstrating the mucosal protective effect of allyphenyline (ALL) and the inhibitory effect of ARC 239 (ARC), (S)-WAY 100135 (WAY), and JP-1302 (JP) on it. The dark, livid areas represent the hemorrhagic ulcerous part of the mucosa
As Fig. 3 shows, the formation of mucosal lesions in the ethanol-treated animals was associated with significantly reduced SOD activity in the gastric mucosa (0.47 ± 0.04 vs 0.32 ± 0.02 unit/mg tissue, n = 5, p < 0.001), while this reduction was completely prevented by allyphenyline (0.53 ± 0.03 unit/mg tissue, n = 5, p < 0.001), indicating that it counteracted the pro-oxidant effect of ethanol. Similarly, the mucosal CAT activity was significantly higher in the allyphenyline + ethanol-treated group than in the ethanol control group (vehicle + ethanol: 0.27 ± 0.03, allyphenyline + ethanol: 0.56 ± 0.15 nmol/min/mg tissue, n = 5–10, p < 0.05).

The effect of ethanol (ETH) and allyphenyline (ALL) on the mucosal catalase (CAT, a) and superoxide dismutase (SOD, b) activity. Allyphenyline (15 nmol) was injected intracerebroventricularly (i.c.v.) 10 min before the ethanol challenge. Each column represents mean (±SEM) of 5–10 rats. *p < 0.05 compared with ethanol-treated group (column 2) (ANOVA, Newman–Keuls post hoc test)
Analyzing the Involvement of α2C-ARs and 5HT1A Receptors in the Gastroprotective Effect of Allyphenyline
The mucosal protective action of i.c.v.-injected allyphenyline was completely inhibited by ARC 239 (50 nmol/rat i.c.v., Fig. 2). Although this compound is widely used as an α2B/C-AR antagonist, there is evidence that it also binds to 5HT1A receptors with nanomolar affinity [37]. Hence, the inhibitory action of ARC 239 indicates only that at least one of these receptors is involved in mediating the effect of allyphenyline. For further analysis, we used (S)-WAY 100135 (50 nmol/rat) and JP-1302 (56 nmol/rat), selective antagonists of 5HT1A receptors and α2C-ARs, respectively [38, 39]. As Fig. 2 shows, both compounds inhibited the effect of allyphenyline, suggesting that both α2C-ARs and 5HT1A receptors contribute to the gastroprotection. When given alone, none of the antagonists influenced significantly the development of ethanol-induced lesions.
The Effect of Allyphenyline on EFS-Induced Fundic and Colonic Contractions
As Fig. 4 demonstrates, allyphenyline inhibited EFS-evoked cholinergic contractions in both fundus and colon in a concentration-dependent fashion. While in the stomach allyphenyline was one order of magnitude less potent than clonidine, a prototypical non-selective α2-AR agonist (EC50 values are 240.8 ± 71.6 and 20.8 ± 6.2 nM, respectively), in the colon they had comparable potencies and efficacies (Table 1). Figure 4b, d shows representative tracings illustrating the effect of allyphenyline on EFS-induced fundic and colonic cholinergic contractions. In contrast to the EFS-evoked contractile response, allyphenyline did not influence the nitrergic relaxation (not shown).

The inhibitory effect of allyphenyline (ALL) and clonidine (CLO) on cholinergic fundic (a) and colonic (c) contractions induced by EFS in C57BL/6 mice. The numbers in brackets indicate the number of experiments. Both agonists were added cumulatively into the organ bath at increasing concentration every 5 min. Each concentration response curve represents the mean ± SEM values of inhibition. b, d Representative tracings illustrating the inhibitory effect of allyphenyline (ALL, 10−9–10−5 M) on the EFS-induced fundic and colonic contractions in C57BL/6 mice
Analyzing the Involvement of α2C-ARs and 5HT1A Receptors in the Allyphenyline-Evoked Inhibition of Fundic and Colonic Contractions
ARC 239 (1 µM) did not influence the amplitude of EFS-induced twitch contractions in the stomach or in the colon per se, but significantly reduced the inhibitory effect of allyphenyline in both organs (Figs. 5a, 6a). However, in the stomach it shifted the concentration–response curve of allyphenyline rightward, and increased its EC50 value without significantly altering its E max, while in the colon it reduced allyphenyline’s E max, without significantly changing its EC50 value (Table 1). These results suggest that the interactions between allyphenyline and ARC 239 in the two organs might involve different mechanisms of action. (S)-WAY 100135 (1 µM), similarly to ARC 239, did not alter EFS-induced fundic and colonic contractions alone, but significantly reduced the effect of allyphenyline in the stomach. On the other hand, it failed to modify that in the colon (Figs. 5b, 6b; Table 1). In contrast to the other two antagonists, JP-1302 (1 µM) affected the effect of allyphenyline neither in the stomach nor in the colon (Figs. 5c, 6c; Table 1).

The antagonistic effect of ARC 239 (ARC, 10−6 M, a), (S)-WAY 100135 (WAY, 10−6 M, b), and JP-1302 (JP, 10−6 M, c) on the inhibitory effect of allyphenyline (ALL, 10−9–10−5 M) on EFS-induced fundic contractions. Each circle represents mean ± SEM values of inhibition (empty circles agonist alone, filled circles: agonist + antagonist), numbers in brackets indicate the number of experiments. # p < 0.05 (two-way repeated measures ANOVA, Holm-Sidak post hoc test)

a–c The antagonistic effect of ARC 239 (ARC, 10−6 M, a), (S)-WAY 100135 (WAY, 10−6 M, b), and JP-1302 (JP, 10−6 M, c) on the inhibitory effect of allyphenyline (ALL, 10−9–10−5 M) on EFS-induced colonic contractions. Each circle represents mean ± SEM values of inhibition (empty circles agonist alone, filled circles agonist + antagonist), numbers in brackets indicate the number of experiments. # p < 0.05 (two-way repeated measures ANOVA, Holm-Sidak post hoc test). d, e The inhibitory effect of allyphenyline (ALL) on EFS-induced colonic contractions in wild-type and α2A- and α2B-adrenoceptor-deficient C57BL/6 mice. Each circle represents mean ± SEM values of inhibition (empty circles wild-type mice, filled circles respective knock out strain), numbers in brackets indicate the number of experiments
These data indicate that 5HT1A receptors are involved in mediating the inhibitory effect of allyphenyline on fundic motility, but neither these receptors nor α2C-ARs take part in the inhibition of EFS-induced contractions in the colon. Though previous in vitro and in vivo studies did not indicate the involvement of α2A- and α2B-ARs in the action of allyphenyline [22–24], we also wanted to exclude the participation of these receptors in the inhibitory effect of allyphenyline on colonic motility. Therefore, we compared its effect in wild-type, α2A- and α2B-AR KO mice. However, as Fig. 6d, e demonstrate, the concentration–response curves were the same in all lines, which rules out the involvement of these α2-AR subtypes in the effect of allyphenyline.
Discussion
In the present study, we aimed to characterize the GI effects of a novel α2C-AR agonist/α2A-AR antagonist/5HT1A receptor agonist agent named allyphenyline, in terms of gastroprotection and regulation of GI motility. Our data demonstrate that allyphenyline possesses powerful gastroprotective action, which is mediated by central α2C-ARs and 5HT1A receptors, and inhibits both fundic and colonic contractions evoked by EFS, although probably via different, 5HT1A receptor-dependent and receptor-independent mechanisms, respectively.
It is well established that peptic ulcers develop as a result of impaired balance between the aggressive and protective factors of gastroduodenal mucosa [40]. Although introduction of antisecretory agents and antibiotic protocols against H. pylori resulted in a significant fall of the incidence of peptic ulcers, there is still a clinical need to find novel gastroprotective drugs which prevent the development and/or accelerate the healing of nonsteroidal anti-inflammatory drug (NSAID)-induced ulcers, H. pylori positive and negative gastroduodenal ulcers as well as stress-related ulcers in critically ill patients [41–43]. Since the historic article of Robert [44] on “gastric cytoprotection,” numerous endogenous and exogenous compounds and receptors have been shown to induce and mediate gastroprotection, i.e., prevention of ulcer formation without influencing gastric acid secretion (recent reviews: [42, 45–49]).
In the last two decades, our group provided mounting evidence for the gastroprotective effect of α2-AR agonists. We demonstrated that clonidine and rilmenidine, when given either peripherally (per os, subcutaneously) or centrally (i.c.v.), inhibited potently the direct necrotizing action of acidified ethanol on gastric mucosa in both rats and mice [12–17]. Our results also showed that the site of action is presumably within the CNS [16, 17], where the activation of α2B- and α2C-ARs initiates a chain of events resulting in vagally mediated release of mucosal protective factors, such as prostaglandins and nitric oxide [13, 16]. Because α2-AR agonists are widely used as antihypertensive drugs, as adjuncts to anesthetics and analgesics during the perioperative period, or as drugs to treat opiate withdrawal or attention-deficit/hyperactivity disorder [50], a potential anti-ulcer effect is of high clinical relevance. However, non-selective activation of all three α2-AR subtypes in the ulcer therapy would result in numerous undesired side effects, mainly mediated by the α2A-AR subtype (hypotension, bradycardia, sedation), but partly also by the α2B-AR subtype (vasoconstriction) [2, 4, 19]. Hence, selective stimulation of α2C-ARs would provide a safer approach to treat upper GI mucosal lesions.
Besides α2-ARs also 5HT1A receptors have been implicated in the regulation of gastric mucosal integrity. Various selective 5HT1A receptor agonists, such as buspiron, 8-OH-DPAT, lesopitron (E-4424), E-4414, or E-4804, given either orally or i.p., were shown to inhibit gastric acid and pepsin secretion, to increase gastric adherent mucus levels and to reduce mucosal injury in both acid-dependent (cold-restraint stress, indomethacin, conditioned fear) and acid-independent (ethanol) ulcer models [27, 28, 51–53]. Whether the site of action is within the CNS or in the periphery has not been established, but Farre et al. [27] (based on unpublished observations) suggested that both central and peripheral effects may contribute to the mucosal protection. The possibility of a central component is indirectly supported by Hoshino and Sugizaki [54], who reported that destruction of the median raphe nucleus, a significant site of central 5-HT cell bodies, was associated with an increased incidence of gastric lesions.
Based on the data described above, we hypothesized that allyphenyline, a novel compound synthetized by Gentili et al. [22] possessing a particularly intriguing receptorial profile (α2C-AR agonist/α2A-AR antagonist/5HT1A receptor agonist), would exert potent gastroprotective action. Our results confirmed this hypothesis and showed that centrally injected allyphenyline almost completely inhibited the development of gastric mucosal damage induced by ethanol, indicating its ability to recruit protective factors and enhance mucosal defense. This effect was inhibited by co-injection with ARC 239 (α2B/C-AR and 5HT1A receptor antagonist), (S)-WAY 100135 (selective 5HT1A receptor antagonist), and JP-1302 (selective α2C-AR antagonist), which suggests that central α2C-ARs and 5HT1A receptors contribute equally to the gastroprotective action. It is of note that similar dual receptorial action was observed by Del Bello et al. [25], who reported that yohimbine and (S)-WAY 100135 similarly contrasted the antidepressive-like effect of allyphenyline.
Although a detailed analysis of the peripheral factors involved in the mucosal protection was above the scope of the present study, we addressed the question of whether allyphenyline is able to counteract oxidative stress, an important factor in the pathomechanism of various experimental ulcers (e.g., NSAID, stress, ethanol) [55–58], as well as in H. pylori-associated ulcers [59]. It is well established that excessive production of reactive oxygen species (ROS) and/or their decreased elimination by antioxidant enzymes induce cell and tissue damage by promoting lipid peroxidation and increasing the production of inflammatory mediators and proinflammatory cytokines [60]. Reduced mucosal antioxidative capacity and increased level of lipid peroxidation products (e.g., malondialdehyde) correlate well with macroscopic and histological mucosal damage, and agents endowed with antioxidant property are able to prevent ROS-mediated cellular damage [55–58, 61]. Our results show that allyphenyline significantly increased the mucosal content of SOD [enzyme which catalyzes the dismutation of O2·− into less noxius hydrogen peroxide (H2O2)] and CAT (which accelerates the breakdown of H2O2 to water and oxygen) compared to the ethanol-treated group, suggesting that the gastroprotective action of allyphenyline—at least partly—relies on its ability to enhance the activity of mucosal antioxidant enzymes and reduce the damaging effect of ROS.
Besides inducing gastric mucosal protection, allyphenyline also inhibited EFS-induced cholinergic contractions in the murine fundus ex vivo. Compared to clonidine, a non-selective α2-AR agent, allyphenyline proved to be one order of magnitude less potent. Because among the three α2-AR subtypes allyphenyline activates only the α2C-AR subtype [22, 23], which does not have any role in the modulation of gastric motility in mice [34], the inhibitory effect of allyphenyline may result from its serotonergic property, and not from an interaction with α2-ARs. Indeed, we found that (S)-WAY 100135 significantly reduced the inhibitory effect of allyphenyline in the stomach, similarly to ARC 239, while JP-1302 had no relevant effect on it. These findings indicate the involvement of 5HT1A receptors in the inhibitory response, which is in line with previous observations. Namely, presynaptic 5-HT1A receptors were shown to mediate the inhibition of transmitter release from cholinergic nerve endings in the guinea pig antrum [29], and various 5HT1A receptor agonists (buspirone, R137696 or flesinoxan) were shown to relax the proximal stomach in mice [30, 62], dogs [63], and also in humans [64, 65]. In accordance with the literature data [63], we found that nitrergic nerves are not involved in the 5-HT1A receptor-mediated fundic relaxation, because allyphenyline did not influence the EFS-induced L-NNA-sensitive relaxation response.
Relaxation of the fundus, either by inhibiting the cholinergic drive or by stimulating the activity of inhibitory (nitrergic or VIP-ergic) neurons, may result in enhanced gastric accommodation [66]. Since impaired gastric accommodation is a hallmark of functional dyspepsia (FD) and contributes to a number of dyspeptic symptoms including nausea and early satiety, improving gastric accommodation by 5HT1A receptor agonists is a promising approach to treat dyspeptic patients [66–68]. Supporting this concept, buspirone and tandospirone significantly improved the symptoms of FD patients [69, 70], although in the case of tandospirone the beneficial effect was attributed mainly to its anxiolytic property. Considering the potent gastroprotective, fundic relaxing and anxiolytic [26] properties of allyphenyline, we propose that this compound may be a useful tool in the therapy of both peptic ulcers and non-ulcer dyspepsia.
Our results demonstrate that in addition to the fundic relaxation, allyphenyline also potently inhibits cholinergic contractions in the colon, however, via an unknown mechanism. First, neither pharmacologic inhibition of α2C-ARs by JP-1302 nor genetic deletion of α2A- and α2B-ARs altered the effect of allyphenyline. These results not only indicate that α2C-ARs are not involved in the control of colonic motility, but also confirm the original findings of Gentili et al. [22] that allyphenyline does not activate the α2A-AR subtype, which otherwise regulates colonic contractility [71]. Second, the effect was not affected by (S)-WAY 100135, which rules out the involvement of 5-HT1A receptors. The exact role of these receptors in the control of colonic motility is still enigmatic [72], but recent findings suggest that these receptors take part in the modulation of colonic migrating motor complex by limiting the neurotransmitter output from intrinsic primary afferent neurons (IPANs) to ascending (S Type I) interneurons [31]. Our results suggest that 5-HT1A receptors are not involved in the inhibition of cholinergic contractions in the murine colon. Interestingly, ARC 239 reduced the colonic effect of allyphenyline, which implies that it interferes with the binding of allyphenyline to another, yet unidentified molecular target. However, even if the exact pharmacodynamic action remains to be established, the modulatory action of allyphenyline in the colon may be useful in various conditions with increased motility. It should be noted that there is a considerable overlap between FD and IBS patients [73, 74], and allyphenyline may have significant impact on most symptoms in these patients.
In summary, we demonstrated herein that allyphenyline possesses powerful gastroprotective action, which is mediated by central α2C-ARs and 5HT1A receptors, and inhibits both fundic and colonic contractions evoked by EFS, via 5HT1A receptor-dependent and receptor-independent mechanisms, respectively. These results (1) confirm the role of central α2C-ARs in the regulation of gastric mucosal integrity and provide the first evidence for the role of central 5HT1A receptors in it, (2) confirm the presence and inhibitory effect of presynaptic 5HT1A receptors on cholinergic motor neurons at the level of stomach, and (3) from a clinical aspect suggest that allyphenyline may be a particularly useful tool in the treatment of various GI disorders, such as peptic ulcers, functional dyspepsia, or irritable bowel syndrome.
References
Bylund DB, Eikenberg DC, Hieble JP, et al. International union of pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;46:121–136.
Hein L. Adrenoceptors and signal transduction in neurons. Cell Tissue Res. 2006;326:541–551.
Knaus AE, Muthig V, Schickinger S, et al. Alpha2-adrenoceptor subtypes-unexpected functions for receptors and ligands derived from gene-targeted mouse models. Neurochem Int. 2007;51:277–281.
Gyires K, Zádori ZS, Török T, Mátyus P. Alpha(2)-adrenoceptor subtypes-mediated physiological, pharmacological actions. Neurochem Int. 2009;55:447–453.
Paton WD, Vizi ES. The inhibitory action of noradrenaline and adrenaline on acetylcholine output by guinea-pig ileum longitudinal muscle strip. Br J Pharmacol. 1969;35:10–28.
De Ponti F, Giaroni C, Cosentino M, Lecchini S, Frigo G. Adrenergic mechanisms in the control of gastrointestinal motility: from basic science to clinical applications. Pharmacol Ther. 1996;69:59–78.
Blandizzi C. Enteric alpha-2 adrenoceptors: pathophysiological implications in functional and inflammatory bowel disorders. Neurochem Int. 2007;51:282–288.
Nagata M, Osumi Y. Central alpha 2-adrenoceptor-mediated inhibition of gastric motility in rats. Jpn J Pharmacol. 1993;62:329–330.
Müllner K, Rónai AZ, Fülöp K, Fürst S, Gyires K. Involvement of central K(ATP) channels in the gastric antisecretory action of alpha2-adrenoceptor agonists and beta-endorphin in rats. Eur J Pharmacol. 2002;435:225–229.
Umezawa T, Guo S, Jiao Y, Hisamitsu T. Effect of clonidine on colonic motility in rats. Auton Neurosci. 2003;107:32–36.
Tack J, Caenepeel P, Corsetti M, Janssens J. Role of tension receptors in dyspeptic patients with hypersensitivity to gastric distention. Gastroenterology. 2004;127:1058–1066.
Fülöp K, Zádori Z, Rónai AZ, Gyires K. Characterisation of alpha2-adrenoceptor subtypes involved in gastric emptying, gastric motility and gastric mucosal defence. Eur J Pharmacol. 2005;528:150–157.
Gyires K, Müllner K, Fürst S, Rónai AZ. Alpha-2 adrenergic and opioid receptor-mediated gastroprotection. J Physiol Paris. 2000;94:117–121.
Gyires K, Müllner K, Rónai AZ. Functional evidence that gastroprotection can be induced by activation of central alpha(2B)-adrenoceptor subtypes in the rat. Eur J Pharmacol. 2000;396:131–135.
Gyires K, Rónai AZ, Müllner K, Fürst S. Intracerebroventricular injection of clonidine releases beta-endorphin to induce mucosal protection in the rat. Neuropharmacology. 2000;39:961–968.
Gyires K, Zádori ZS, Shujaa N, Minorics R, Falkay G, Mátyus P. Analysis of the role of central and peripheral alpha2-adrenoceptor subtypes in gastric mucosal defense in the rat. Neurochem Int. 2007;51:289–296.
Zádori ZS, Shujaa N, Brancati SB, Hein L, Gyires K. Both alpha2B- and alpha2C-adrenoceptor subtypes are involved in the mediation of centrally induced gastroprotection in mice. Eur J Pharmacol. 2011;669:115–120.
Docherty JR. Subtypes of functional alpha1- and alpha2-adrenoceptors. Eur J Pharmacol. 1998;361:1–15.
Philipp M, Brede M, Hein L. Physiological significance of alpha(2)-adrenergic receptor subtype diversity: one receptor is not enough. Am J Physiol Regul Integr Comp Physiol. 2002;283:287–295.
Link RE, Desai K, Hein L, et al. Cardiovascular regulation in mice lacking alpha2-adrenergic receptor subtypes b and c. Science. 1996;273:803–805.
Makaritsis KP, Johns C, Gavras I, et al. Sympathoinhibitory function of the alpha(2A)-adrenergic receptor subtype. Hypertension. 1999;34:403–407.
Gentili F, Cardinaletti C, Vesprini C, et al. Alpha2-adrenoreceptors profile modulation. 4. From antagonist to agonist behavior. J Med Chem. 2008;51:4289–4299.
Cardinaletti C, Mattioli L, Ghelfi F, et al. Might adrenergic alpha2C-agonists/alpha2A-antagonists become novel therapeutic tools for pain treatment with morphine? J Med Chem. 2009;52:7319–7322.
Del Bello F, Mattioli L, Ghelfi F, et al. Fruitful adrenergic alpha(2C)-agonism/alpha(2A)-antagonism combination to prevent and contrast morphine tolerance and dependence. J Med Chem. 2010;53:7825–7835.
Del Bello F, Diamanti E, Giannella M, et al. Low doses of allyphenyline and cyclomethyline, effective against morphine dependence, elicit an antidepressant-like effect. ACS Med Chem Lett. 2012;3:535–539.
Ubaldi M, Del Bello F, Domi E, Pigini M, Nasuti C. Investigation of allyphenyline efficacy in the treatment of alcohol withdrawal symptoms. Eur J Pharmacol. 2015;760:122–128.
Farre AJ, Colombo M, Alvarez I, Glavin GB. Some novel 5-hydroxytryptamine1A (5-HT1A) receptor agonists reduce gastric acid and pepsin secretion, reduce experimental gastric mucosal injury and enhance gastric mucus in rats. J Pharmacol Exp Ther. 1995;272:832–837.
Abdel Salam O, Baiuomy A. Effect of buspirone on inflammation, pain and gastric injury in mice. Int J Pharmacol. 2007;6.
Tack JF, Janssens J, Vantrappen G, Wood JD. Actions of 5-hydroxytryptamine on myenteric neurons in guinea pig gastric antrum. Am J Physiol. 1992;263:838–846.
Xue L, Camilleri M, Locke GR 3rd, et al. Serotonergic modulation of murine fundic tone. Am J Physiol Gastrointest Liver Physiol. 2006;291:1180–1186.
Dickson EJ, Heredia DJ, Smith TK. Critical role of 5-HT1A, 5-HT3, and 5-HT7 receptor subtypes in the initiation, generation, and propagation of the murine colonic migrating motor complex. Am J Physiol Gastrointest Liver Physiol. 2010;299:144–157.
Takeuchi K. Pathogenesis of NSAID-induced gastric damage: importance of cyclooxygenase inhibition and gastric hypermotility. World J Gastroenterol. 2012;18:2147–2160.
Voronina TA, Glozman OM, Orlova EK, et al. Synthesis and psychotropic activity of 2-phenoxypropionic oxamidines and their analogs. Khim Farm Z. 1984;18:1309–1313.
Shujaa N, Al-Khrasani M, Zádori ZS, et al. alpha(2)-adrenoceptor agonist-induced inhibition of gastric motor activity is mediated by alpha(2A)-adrenoceptor subtype in the mouse. Neurochem Int. 2011;58:708–713.
Altman JD, Trendelenburg AU, MacMillan L, et al. Abnormal regulation of the sympathetic nervous system in alpha2A-adrenergic receptor knockout mice. Mol Pharmacol. 1999;56:154–161.
Philipp M, Brede ME, Hadamek K, Gessler M, Lohse MJ, Hein L. Placental alpha(2)-adrenoceptors control vascular development at the interface between mother and embryo. Nat Genet. 2002;31:311–315.
Meana JJ, Callado LF, Pazos A, Grijalba B, Garcia-Sevilla JA. The subtype-selective alpha 2-adrenoceptor antagonists BRL 44408 and ARC 239 also recognize 5-HT1A receptors in the rat brain. Eur J Pharmacol. 1996;312:385–388.
Fletcher A, Bill DJ, Bill SJ, et al. WAY100135: a novel, selective antagonist at presynaptic and postsynaptic 5-HT1A receptors. Eur J Pharmacol. 1993;237:283–291.
Sallinen J, Hoglund I, Engstrom M, et al. Pharmacological characterization and CNS effects of a novel highly selective alpha2C-adrenoceptor antagonist JP-1302. Br J Pharmacol. 2007;150:391–402.
Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology. 2008;135:41–60.
Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet. 2009;374:1449–1461.
Szabo S. “Gastric cytoprotection” is still relevant. J Gastroenterol Hepatol. 2014;29:124–132.
Bardou M, Quenot JP, Barkun A. Stress-related mucosal disease in the critically ill patient. Nat Rev Gastroenterol Hepatol. 2015;12:98–107.
Robert A, Nezamis JE, Lancaster C, Hanchar AJ. Cytoprotection by prostaglandins in rats. Prevention of gastric necrosis produced by alcohol, HCl, NaOH, hypertonic NaCl, and thermal injury. Gastroenterology. 1979;77:433–443.
Tache Y. Brainstem neuropeptides and vagal protection of the gastric mucosal against injury: role of prostaglandins, nitric oxide and calcitonin-gene related peptide in capsaicin afferents. Curr Med Chem. 2012;19:35–42.
Kemmerly T, Kaunitz JD. Gastroduodenal mucosal defense. Curr Opin Gastroenterol. 2013;29:642–649.
Gyires K, Németh J, Zádori ZS. Gastric mucosal protection and central nervous system. Curr Pharm Des. 2013;19:34–39.
Gyires K, Zádori ZS. Brain neuropeptides in gastric mucosal protection. Curr Opin Pharmacol. 2014;19:24–30.
Takeuchi K. Gastric cytoprotection by prostaglandin E(2) and prostacyclin: relationship to EP1 and IP receptors. J Physiol Pharmacol. 2014;65:3–14.
Crassous PA, Denis C, Paris H, Senard JM. Interest of alpha2-adrenergic agonists and antagonists in clinical practice: background, facts and perspectives. Curr Top Med Chem. 2007;7:187–194.
Sullivan RM, Henke PG, Ray A. The effects of buspirone, a selective anxiolytic, on stress ulcer formation in rats. Pharmacol Biochem Behav. 1988;31:317–319.
Glavin GB, Alvarez I, Colombo M, Farre AJ. Effects of a novel 5-HT1A receptor agonist, E4424, on gastric adherent mucus levels following restraint stress in rats. Dig Dis Sci. 1995;40:2317–2320.
Krysiak R, Obuchowicz E, Herman ZS. Conditioned fear-induced changes in neuropeptide Y-like immunoreactivity in rats: the effect of diazepam and buspirone. Neuropeptides. 2000;34:148–157.
Hoshino K, Sugizaki M. Ulcerogenic effect of the lesion of the median raphe nucleus in fasted rats. Braz J Med Biol Res. 1986;19:123–130.
Yoshikawa T, Naito Y, Kishi A, et al. Role of active oxygen, lipid peroxidation, and antioxidants in the pathogenesis of gastric mucosal injury induced by indomethacin in rats. Gut. 1993;34:732–737.
Kim SJ, Park YS, Paik HD, Chang HI. Effect of anthocyanins on expression of matrix metalloproteinase-2 in naproxen-induced gastric ulcers. Br J Nutr. 2011;106:1792–1801.
Kwiecien S, Brzozowski T, Konturek PC, et al. The role of reactive oxygen species and capsaicin-sensitive sensory nerves in the pathomechanisms of gastric ulcers induced by stress. J Physiol Pharmacol. 2003;54:423–437.
Sangiovanni E, Vrhovsek U, Rossoni G, et al. Ellagitannins from Rubus berries for the control of gastric inflammation: in vitro and in vivo studies. PLoS ONE. 2013;8:e71762.
Davies GR, Simmonds NJ, Stevens TR, et al. Helicobacter pylori stimulates antral mucosal reactive oxygen metabolite production in vivo. Gut. 1994;35:179–185.
Kwiecien S, Jasnos K, Magierowski M, et al. Lipid peroxidation, reactive oxygen species and antioxidative factors in the pathogenesis of gastric mucosal lesions and mechanism of protection against oxidative stress - induced gastric injury. J Physiol Pharmacol. 2014;65:613–622.
El-Maraghy SA, Rizk SM, Shahin NN. Gastroprotective effect of crocin in ethanol-induced gastric injury in rats. Chem Biol Interact. 2015;229:26–35.
Xue L, Locke GR, Camilleri M, et al. Effect of modulation of serotonergic, cholinergic, and nitrergic pathways on murine fundic size and compliance measured by ultrasonomicrometry. Am J Physiol Gastrointest Liver Physiol. 2006;290:74–82.
Janssen P, Prins NH, Moreaux B, Meulemans AL, Lefebvre RA. In vivo characterization of 5-HT1A receptor-mediated gastric relaxation in conscious dogs. Br J Pharmacol. 2003;140:913–920.
Tack J, Piessevaux H, Coulie B, Fischler B, De Gucht V, Jannsens J. A placebo-controlled trial of buspirone, a fundus-relaxing drug, in functional dyspepsia: effect on symptoms and gastric sensory and motor function. Gastroenterology. 1999;166:325.
Boeckxstaens GE, Tytgat GN, Wajs E, et al. The influence of the novel 5-HT1A agonist R137696 on the proximal stomach function in healthy volunteers. Neurogastroenterol Motil. 2006;18:919–926.
Tack J. Prokinetics and fundic relaxants in upper functional GI disorders. Curr Opin Pharmacol. 2008;8:690–696.
Tack J, Demedts I, Meulemans A, Schuurkes J, Janssens J. Role of nitric oxide in the gastric accommodation reflex and in meal induced satiety in humans. Gut. 2002;51:219–224.
Van Oudenhove L, Kindt S, Vos R, Coulie B, Tack J. Influence of buspirone on gastric sensorimotor function in man. Aliment Pharmacol Ther. 2008;28:1326–1333.
Tack J, Janssen P, Masaoka T, Farre R, Van Oudenhove L. Efficacy of buspirone, a fundus-relaxing drug, in patients with functional dyspepsia. Clin Gastroenterol Hepatol. 2012;10:1239–1245.
Miwa H, Nagahara A, Tominaga K, et al. Efficacy of the 5-HT1A agonist tandospirone citrate in improving symptoms of patients with functional dyspepsia: a randomized controlled trial. Am J Gastroenterol. 2009;104:2779–2787.
Zhang L, Keef KD, Bradley ME, Buxton IL. Action of alpha 2A-adrenergic receptors in circular smooth muscle of canine proximal colon. Am J Physiol. 1992;262:517–524.
Smith TK, Park KJ, Hennig GW. Colonic migrating motor complexes, high amplitude propagating contractions, neural reflexes and the importance of neuronal and mucosal serotonin. J Neurogastroenterol Motil. 2014;20:423–446.
Caballero-Plasencia AM, Sofos-Kontoyannis S, Valenzuela-Barranco M, Martin-Ruiz JL, Casado-Caballero FJ, Lopez-Manas JG. Irritable bowel syndrome in patients with dyspepsia: a community-based study in southern Europe. Eur J Gastroenterol Hepatol. 1999;11:517–522.
Kaji M, Fujiwara Y, Shiba M, et al. Prevalence of overlaps between GERD, FD and IBS and impact on health-related quality of life. J Gastroenterol Hepatol. 2010;25:1151–1156.
Acknowledgments
Our research was supported by the Hungarian Scientific Research Fund (OTKA PD 109602). The authors wish to thank Mrs. I. Szalai for her technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Additional information
Zoltán S. Zádori and Ágnes Fehér have equally contributed to this work.
Rights and permissions
About this article

Cite this article
Zádori, Z.S., Fehér, Á., Tóth, V.E. et al. Dual Alpha2C/5HT1A Receptor Agonist Allyphenyline Induces Gastroprotection and Inhibits Fundic and Colonic Contractility. Dig Dis Sci 61, 1512–1523 (2016). https://doi.org/10.1007/s10620-015-4026-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-015-4026-9