
Abstract
Cellular radioresistance is one of the major obstacles to the effectiveness of cancer radiotherapy. In an attempt to elucidate the implication of HIF-1α and miR-17-92 expressions in refractory radioresistant cells and also in order to study the potential applications of these molecules as novel therapeutic modalities to overcome radioresistant cancers, the current study was conducted. Clinically relevant radioresistant (CRR) cells from human cancer cell lines were established by exposing to long-term fractionated radiation of X-rays. Correspondingly, microarray analysis and real time RT-PCR were performed to find miRNA involved in the CRR phenotype. HIF-1α was down-regulated and miR17-92 cluster was overexpressed in CRR cells by transfection. The expression of miR 17-3p was inhibited by specific inhibitors and miR 19a was enforced by mimics, respectively in parental cells. Overexpression of HIF-1α in parental cells or down regulation of HIF-1α in CRR cells were not involved in radioresistance. However, when HIF-1α was genetically modified to constitutively express under normoxia condition, it was rendered for protection to cells. Exogenous overexpression of miR 17-92 cluster in CRR cells resulted in abolition of HIF-1α expression and restored sensitizations to ionizing radiation. Attenuated expression of miR-17-3p in parental cells protected them from irradiation. Overall, fine-tune deregulation of miR 17-92 cluster in CRR cells might account for the accumulation of HIF-1α in the CRR cells following exposure to irradiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Radiotherapy is one of the major modalities for cancer therapy. Recent advances in physical targeting of ionizing radiation (IR), optimization of IR delivery schedules, and tumor imaging have resulted in considerable improvements in the patient outcomes (Schaue and McBride 2015). However, the existence of cellular radioresistance is one of the major impediment to the efficiency and versatility of radiotherapy (RT) (Kuwahara et al. 2017, 2018; Shimura 2011; Tomita et al. 2018). Radioresistance is influenced by various intracellular and extracellular factors. MicroRNAs (miRNAs) are noncoding RNAs, approximately 22 nucleotides in length, which function as post-transcriptional regulators (Cellini et al. 2014; Schoof et al. 2012; Zhao et al. 2013; Zhou et al. 2015) by controlling mRNA stability and translation efficiency (Guo et al. 2010; Ye et al. 2012).
Hypoxia, an oxygen concentration below physiological levels, is a well-known influential extracellular factor. Conspicuously, hypoxia disrupts the production of free radicals and thereby interferes with the beneficial effects of radiotherapy. Hypoxia-inducible factor-1(HIF-1) is a heterodimeric protein, consisting of α and β subunits and the HIF-1α subunit is regulated by O2 concentration (Poon et al. 2009; Vaupel and Mayer 2007). Normoxia causes rapid degradation of HIF-1α, whereas hypoxia stabilizes it. Activation of the PI3K/AKT/mTOR pathway and reactive oxygen species inhibit proteasomal degradation of HIF-1α (Ladelfa et al. 2011; Ranasinghe et al. 2014) resulting in its accumulation.
It is known that upregulation and activity of HIF-1α have been implicated in many cancers and correlated with a poor prognosis and tumor recurrence after RT (Ranasinghe et al. 2014). However, the role of HIF-1α in radiation resistance in cancer cells is controversial (Vordermark et al. 2004; Arvold et al. 2005; Staab et al. 2011). Furthermore, there are no studies that indicate the direct implication of HIF-1α in radioresistance phenotype in vitro under normoxic conditions.
Interestingly, both HIF-1α and the miRNA-17-92 cluster target multiple cellular pathways favouring tumorigenesis by enhancing cell proliferation and inhibiting apoptosis suggesting HIF-1α and the miRNA-17-92 cluster may be involved in common biological functions (Taguchi et al. 2008). Moreover, functional annotation indicated that target genes of the miR-17-92 cluster are involved in the regulation of radiation associated signal pathways (Mendell 2008; Taguchi et al. 2008). It has also been reported that the upregulation of miR-17-3p remarkably sensitized PCa cells to ionizing radiation (IR) (Xu et al. 2018). Several studies have been reported to show the implication of HIF-1α in radioresistance (Moeller et al. 2004; Vordermark et al. 2004). Nevertheless, the implication of HIF-1α and miR-17-92 cluster in radio-resistance response remains to be investigated.
Conventional fractionated RT consists of 2 Gy per fraction once a day, 5 days a week for 5–7 weeks (Shimura 2011). To elucidate the molecular mechanisms of cellular radioresistance we have established clinically relevant radioresistant (CRR) cell lines from several human cancer cell lines by long-term exposure to X-rays with a stepwise dose escalation (Kuwahara et al. 2011, 2018; Shimura 2011).
CRR cells continue to proliferate under exposures to fractionated radiation (FR) of 2 Gy/day of X-rays for more than 30 days, a standard protocol of cancer radiotherapy (Kuwahara et al. 2011). CRR cells are an ideal model to better understand the molecular mechanisms behind cellular radioresistance. The present study was conducted to elucidate the implication of HIF-1α and miR-17-92 expressions in refractory radioresistant cells and to investigate potential applications of these molecules as new therapeutic modalities to overcome radioresistant cancers.
Materials and methods
Plasmid DNA
Plasmids pcDNA4/myc-His and pcDNA3.1 were purchased from Invitrogen (Carlsbad, CA, USA). HIF-1α was cloned into pcDNA4/myc or pcDNA3.1 as described previously (Kiani et al. 2013). Constitutively HIF-1α expressing plasmid under normoxia i.e. pcDNA4/myc-His-HIF-1α-3A were constructed by introducing three mutations of alanine instead of the wild-type residues of proline 402, proline 564 and asparagine 803. HIF-1α expressed from HIF-1α-3A construct is stable under normoxia condition. pcDNA3.1CMVpuro-miR17-92 was kindly gifted by Dr. Takahashi, Nagoya University. pGFP-V-RS-HIF-1α shRNAs and pGFP-V-RS-negative non-effective shRNA were purchased from Origen (OriGene, USA).
Cell culture, reagents, and treatments
Human cancer cell lines, SAS (oral squamous cell carcinoma), HepG2 (liver cancer) and HeLa (cervical cancer cell line) were obtained from the Cell Resource Center for Biomedical Research, Institute of development, Aging, and Cancer, Tohoku University. HeLa-5HRE-DsRed2 cells containing reporter gene under the promoter of HIF-1α, which emit red fluorescence under hypoxic conditions in response to HIF-1 activity (Harada et al. 2005).
All cell lines were cultured in Roswell Park Memorial Institute 1640 medium (RPMI-1640) (Nacalai Tesuque, Kyoto, Japan) and supplemented with 5% fetal bovine serum (Invitrogen) in a humidified atmosphere at 37 °C in the air with 5% CO2. For cultivation of cells under hypoxia condition they were underwent 1% O2 condition in a multi-gas incubator Prescyto MG70M (Saitama-ken, Japan).
CRR cell lines, SAS-R1, HepG2-8960-R and HeLa-R were established by exposing them to FR of X-rays for more than 5 years as described previously (Kuwahara et al. 2011, 2017; Shimura 2011). The plasmid vectors were transfected to cells using either FuGENE HD (Promega, Madison, WI, USA) or Lipofectamine 2000 (Invitrogen) according to manufactures’ protocols. The stable clones were selected in the presence of appropriate antibiotics, 450 µg/ml G418 (Sigma, St Louis, MO, USA), 1.5 µg/ml puromycin (InvivoGen, San Diego, CA, USA) or 150 µg/ml Zeocin (Invitrogen). Synthetic inhibitors of miR 19a, miR17-3p, synthetic mimics of miR19a, miR17-p and the scramble controls were obtained from Dharmacon and were transfected with DharmFECT1 (Dharmacon, USA) at a final concentration of 60 nM.
Irradiation
X-ray irradiation was performed in a 150 KVp X-ray generator (Model MBR-1520R, Hitachi, Tokyo, Japan) with a total filtration of 0.5 mm aluminum plus 0.1 mm copper filter, at a dose rate of 1.0-Gy/min.
Cell survival assay after irradiation
Cell survival was determined by the modified high-density survival (MHDS) assay as described previously (Kuwahara et al. 2010). In some experiments, cell survival was determined by colony formation assays following irradiation as described (Feng et al. 2014).
Microarray analysis
The samples of SAS-R and HepG2-R and their parental counterparts were processed for total RNA extraction with the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocols. For Quality control of extracted RNA, the samples were electrophoresed on agarose gel (2%) and for quantity measurement, the extracted RNA was checked by NanoDrop®-1000 Detector (NanoDrop-Technologies, Wilmington, USA). Next, the samples were labeled using a miRCURY Hy3/Hy5 Power labeling kit and followed by hybridization on a 3D-Gene™ Toray miRNA Oligo chip (v.13.0; Toray Industries, Tokyo, Japan). 3D-Gene Scanner 3000 was used for scanning (Toray Industries). To read the raw intensity of the image 3D-Gene extraction version 1.2 software was used (Toray Industries). The raw data were analyzed via GeneSpringGX v 10.0 (Agilent Technologies) in order to determine the change in miRNA expression between the parental and their CRR counterpart cells. The samples were normalized relative to 28sRNA.
Western blot analysis
Western blot of whole cell lysates was performed as previously described (Shimura et al. 2010).
Briefly, the cells were lysed in a lysis buffer [0.5 M NaCl, 1 mM EDTA, 25 mM sodium phosphate buffer pH 7.4, 10% glycerol, 5 mM MgCl2, 1 mM dichlorodiphenyltrichloroethane, 0.5% Triton X-100, 1 mM PMSF, protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan) and phosphatase inhibitor cocktail (Nacalai Tesque)]. Proteins were separated by 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred electrophoretically to PVDF membranes (Bio-Rad, Hercules, CA, USA). 5% (w/v) skim-milk was used to block the membranes (Cell Biolabs, San Diego, CA, USA) for 1 h. Then, the membranes were incubated with primary antibodies overnight at 4 °C. Membranes were then incubated for 1 h at room temperature with the secondary antibodies. The protein bands were observed by Chemi-Lumi One L western blotting substrate (Nacalai Tesque).
The primary antibodies in the present study were anti-β-actin (A5316, Sigma), anti-HIF-1α (NB100-105, Novus Biologicals, Littleton, CO, USA) and the secondary antibodies were goat anti-rabbit IgG (H1202, Nichirei Bioscience, Tokyo, Japan) and mouse anti-rat IgG (H1104, Nichirei Bioscience).
Reverse transcription (RT)–polymerase chain reaction (PCR)
Total RNAs were extracted using a miRNeasy Mini Kit (Qiagen, Valencia, CA, and USA). Relatively, cDNA was synthesized by the miScript Reverse Transcription Kit (Qiagen). RT-PCR was performed using a primer pair, miR-19a_1, miR17-3p and Hs_RNU6-2_1 miScript Primer Assay (Qiagen). Real-time RT-PCR was performed using miScript SYBR Green PCR Kit (Qiagen). After an initial denaturation at 95 °C for 15 min, cDNA was subjected to 40 cycles of 94 °C for 15 s, 55 °C for 30 s and 70 °C for 30 s using Thermal Cycler Dice Real-Time System (Takara, Shiga, Japan).
Statistical analysis
Data are expressed as mean ± SD. Independent experiments were performed to evaluate significant differences between control and other experimental groups. Significant differences were determined by one-way ANOVA with post-hoc Tukey test.
Results
Parental and their CRR counterpart cells differently express HIF-1α upon irradiation
To investigate whether HIF-1α is implicated in radioresistance, first, we established a CRR cell line containing reporter for HIF-1α expression, HeLa-R-5HRE-DsRed2. Then, these cells and their parental counterparts were cultured under normoxic and hypoxic conditions. As we expected, both HeLa-5HRE-DsRed and HeLa-R-5HRE-DsRed2 responded similarly to hypoxia in terms of induction of reporter gene via HIF-1α activation (Fig. 1). However and interestingly, under normoxic conditions, the reporter gene was also expressed in HeLa-R (Fig. 1). To compare the responses of HIF-1α expression upon irradiation, the time-course expression of HIF-1α after irradiation was studied by western blot analysis. HIF-1α expression was somewhat refractory and was slightly decreased after 24 h in the parental cells. However, in the CRR cells it was kept up-regulated (Fig. 2). These results suggest the contribution of HIF-1α in the radioresistance property of CRR cells and indicate that the accumulation of HIF-1α in parental and CRR cells is differently regulated.

Detection of reporter gene indicating of HIF-1α expression parental and CRR cells by florescent microscope. HeLa-5HRE-DsRed2 and its CRR corresponding parent i.e. HeLa-R-5HRE-DsRed2 were cultured under normoxic and hypoxic conditions. Under hypoxia condition, both parental and CRR cells expressed reporter gene while under normoxic conditions only it was observed in CRR cells. Schematic representations of the reporter gene that is regulated under control of HIF-1α promoter and when it activates and inactivates have also been shown in the top of the figure

Study of HIF-1α expression in the parental and CRR cells by western blot analysis. Parental (HepG2, HeLa, and SAS) and CRR cells (HepG2-8960-R, HeLa-R, and SAS-R) were underwent 10-Gy irradiation followed by evaluation of HIF-1α expression at different time points. Expression of HIF-1α was stabilized in CRR cells after 12 h while in the parental cells was decreased at this time. β-actin was also detected as loading a control. NIOR the cells with no irradiation, h hours, D days, IR irradiation
HIF-1α that stable upon normoxic condition protects cells from irradiation
Next, we stably overexpressed HIF-1α in HeLa, SAS and HepG2 (Fig. 3a, b). Then, these cells were irradiated under normoxic conditions followed by cell survival assay. Overexpression of wild type HIF-1α resulted in marginal but not significant protection to irradiation (Fig. 3c). However, when the cells stably overexpressing so-called HIF-1α-3A (Fig. 3b), HIF-1α was permanently expressed under normoxic condition, and exposed to irradiation the survival fractions were higher than those transfected with empty vector (Fig. 3c).

Overexpression of HIF-1α in the parental cells and study of their radioresistance. a Western blot analysis for detection of HIF-1α overexpression in Hela, SAS, and HepG2. The cells were transfected with an empty vector (EV), that containing HIF-1α or HIF-1α-3A. β-Actin was used for internal control. b Cells stably transfected with HIF-1α-3A underwent normoxic and hypoxic conditions. Under hypoxic HIF-1α expression was induced in control cells transfected with an empty vector. However, a high level of expression of HIF-1α was observed in the cells transfected with the 16 construct containing HIF-1α -3A under normoxia condition as well. β-actin was used as loading control. c Survival assay of the parental cells in which HIF-1α or HIF1α -3A was overexpressed as described above. HIF-1α -3A protected cells against irradiation. CI, HepG2; CII, SAS; CIII, Hela; EV, empty vector; − , normoxia; +, hypoxia (mean ± SD, number of samples = 3, **p ≤ 0.01)
Downregulation of HIF-1α in CRR cells and their sensitization response to irradiation
Next, we determined whether the down-regulation of HIF-1α (Fig. 4a) results in the sensitization of CRR cells to irradiation or not. HIF-1α downregulation failed to sensitize of CRR cells to irradiation except for HepG2 8960-R (Fig. 4b).

Downregulation of HIF-1α in CRR cells and survival assay. a Western blot analysis for detection of down regulation of HIF-1α in Hela-R, HepG2-8960-R, and SAS-R1 cells. β-actin was used as an internal control. B; Survival assay in CRR cells transfected with HIF-1α shRNA or control shRNA. Cells exposed to different doses of irradiation. Downregulation of HIF-1α had no significant effects in the CRR cells in terms of sensitization to irradiation except HepG2 (mean ± SD, number of samples = 3, *p ≤ 0.5)
miR-17-92 cluster downregulates in CRR cells
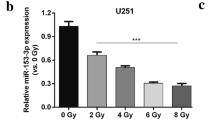
The profile of HIF-1α induction both in CRR and parental cells was similar to the report by Taguchi et al. that HIF-1α is induced by hypoxia regardless of miR-17-92 expression but under normoxic condition miR-17-92 negatively regulates HIF-1α expression (Taguchi et al. 2008). Microarray analysis was performed to see the association of miR-17-92 expression is different or not between CRR cells and parental cells. Except for miR-92, the miR-17-92 cluster was downregulated in CRR cells, (considerably miR-19a and 17-3p) 12 h after exposure to 2 Gy compared with the parental counterparts (Fig. 5 and Table 1). Real-time (RT)-PCR analysis confirmed the downregulation of miR-19a and 17-3p (Supplementary Fig. 1a, b). These suggest that HIF-1α overexpression in CRR cells is attributable to the downregulation of the miR-17-92 cluster.

Microarray analysis of miRNA cluster in CRR cells. Unsupervised hierarchical clustering analysis indicates downregulation of miRNA 17-92 cluster. SAS and HepG2 and their counterpart CRR cells were exposed to 2 Gy X-rays and searched for miR17-92 cluster with an altered expression between the parental and their CRR cells. miRNA 17-92 cluster was down regulated in CRR cells
Overexpression of miR-17-92 cluster in CRR cells abolished the induction of HIF-1α after irradiation
To attribute HIF-1α overexpression in CRR cells following irradiation could be due to downregulation of miR-17-92 cluster, CRR cells with stable overexpression of the miR-17-92 cluster were established by transfecting miR-17-92 cluster to CRR cells (Supplementary Fig. 2).
The response of HIF-1α expression in the CRR cells with stable overexpression of miR-17-92 response was studied by western blot analysis after exposure to 10 Gy of X-rays. Interestingly miR-17-92 overexpression in CRR cells disturbed the induction of HIF1-α (Fig. 6a). These indicate that miR-17-92 negatively regulates HIF-1α expression in the CRR cells.

Overexpression of miR17-92 in the CRR cells and survival assay. a Western blot analysis for detection of HIF-1α expression. CRR cells were transfected with empty vector and the vector containing miR17-92 cluster. Then, the stable cells exposed to 10 Gy of irradiation and expression of HIF-1α at different time point was evaluated. Exogenous expression of miR17-92 prevents the stabilization of HIF-1α in comparisons with CRR cells transfected with an empty vector. β-Actin was used for control. b Cell survival assay in CRR cells overexpression miR17-92 cluster. The CRR cells were exposed to different doses of irradiation followed by cell survival assay. The overexpression resulted in sensitization of CRR cells to irradiation. IR Irradiation (mean ± SD, number of samples = 3, *p ≤ 0.5, **p ≤ 0.01, ***p ≤ 0.001)
Overexpression of miRNA-17-92 in CRR cells restore sensitivity of the acquired radioresistance cells to ionizing radiation
In order to determine whether miRNA-17-92 overexpression would affect the sensitivity of CRR cells to irradiation or not survival assay was performed. Overexpression of miRNA-17-92 significantly decreased the survival of CRR cells after irradiation (Fig. 6b). However, overexpression of miRNA-17-92 in the parental cells had marginal or no effects in terms of cell survival following irradiation (Supplementary Fig. 3).
Effects of miR19a and 17-3p inhibition or overexpression on the parental cells
As described above, miR19 and 17-3p were notably downregulated in CRR cells. On the other hand, next we investigated the effect of inhibition or induction of miR 19a and 17-3p in parental cells. They were down regulated or overexpressed by the synthetic inhibitors or inducers (mimics) (Supplementary Fig. 4a, b), exposed to irradiation and followed by survival assay. Inhibition of miR19a resulted in sensitization of parental cells to irradiation (Fig. 7a. Interestingly, inhibition of 17-3p protected cells to irradiation (Fig. 7b). Overexpression of miR-17-3p slightly sensitized HeLa and SAS but protected HepG2 cells. Overexpression of miR19a protected HeLa and HepG2 and had no effect on SAS cells to irradiation (Supplementary Fig. 5a, b). Altogether, although fine tune regulation of miR17-92 cluster is essential to determine sensitization or resistance of cells to irradiation, down regulation of miR17-3p plays an important role to confer radioresistance.

Cell survival assay in HeLa, SAS and HepG2 cells after attenuating expression of miR-19a and miR17-3p. a miR-19a or miR17-3p were attenuated in cells, exposed to different doses of irradiation followed. Repression of miR-19a expression resulted in more sensitization to irradiation. b Repression of miR17-3p expression resulted in refractory to irradiation. Scr scramble inhibitor or mimic control. G Grays (mean ± SD, number of samples = 3, *p ≤ 0.5, **p ≤ 0.01, ***p ≤ 0.000)
Discussion
Our CRR cells and their isogenic parental cells are suitable to elucidate the molecular mechanisms truly involved in radioresistance (Kuwahara et al. 2017, 2018). A number of studies have reported that HIF-1α plays an important role in radioresistance of cancer cells. However, as far as we are aware, this is the first study that tries to find the relationship between HIF-1α and miR17-92 cluster in radioresistance of cancer cells. In this study, HIF-1α expression was higher in CRR cells than their corresponding parental cells under normoxia, suggesting that HIF-1α might play a role in the CRR phenotype.
It is well-known that oxygen can enhance the cytotoxic effect of radiation (Patterson et al. 2002). However under hypoxic conditions, ionized DNA is more easily repaired and the enhancement effect would be abolished (Sprong et al. 2006). In addition, radiation activates HIF-1a even under normoxic conditions (Zhang et al. 2015). Our result showed that overexpression of HIF-1α per se is not sufficient to confer radioresistance to cells. However constant HIF-1α overexpression protected cells from radiation-induced cell death even under normoxic conditions. In line with our findings, it has been shown that pre-treatment of HeLa cells in hypoxic conditions resulted in increased cellular radioresistance (Fu et al. 2015). Recently, Doi et al. reported that hypoxia mimicked by cobalt chloride enhanced radioresistance in HeLa cells (Doi et al. 2015). Of note, cobalt chloride had no effect on the oxygenation of cells but activates the hypoxia signaling transduction pathway, especially the HIF-1α pathway (Liu et al. 2010). Nevertheless, controversial reports have been also reported to address the role of HIF-1 α in radiation resistance in cancer cells both in vivo and in vitro. Vordemark et al. have shown that the HIF-1α protein accumulation levels under hypoxia do not always correlate with cellular radioresistance (Vordermark and Katzer 2004). Arvold et al. demonstrated that the radiosensitivity of mouse embryonic fibroblasts (MEF) following exposure to hypoxia was independent of HIF-1α (Arvold and Guha 2005). Although the manipulation of HIF-1α did not affect radiosensitivity in fibrosarcoma cells under normoxic conditions (Staab et al. 2011) but overexpression of HIF-1α and culture under hypoxia conditions increased radioresistance in HeLa cells (Liu et al. 2010).
It has been reported that there is an association between HIF-1α overexpression in tumors with poor clinical outcomes and radioresistance (Aebersold et al. 2001; Dellas et al. 2008; Koukourakis et al. 2002; Nordsmark et al. 2007; Semenza 2010). Hence, we also down-regulated the expression of HIF-1α in CRR cells intending to sensitize them to irradiation. However, our results revealed that down regulation of HIF-1α had marginal or no effects on radioresistance of CRR cells.
It could be possible that HIF-2α complement HIF-1α. Supporting this notion, Doi et al. investigated the effect of knockdown of both HIF-1α and HIF-2α on HeLa cells. After exposing to simulated hypoxia, they found that radioresistance of HeLa cells decreased more than in either single HIF gene knockdown, suggesting that both genes are essential to acquire radioresistance (Doi et al. 2015). Further investigations on CRR cells, including potential mechanisms, are required to see contribution both HIF-1α and HIF-2α in radioresistance.
According to the literature, two non-cell autonomous and autonomous mechanisms have been proposed to explain the implication of HIF-1α in radioresistance. Based upon the non-cell autonomous mechanism, radiation activates HIF-1α, then it induces vascular endothelial growth factor (VEGF) secretion which protects endothelial cells from radiation-induced cell death promotes ultimately tumor growth (Harada 2011). Activation of HIF-1α also results in up-regulation of SDF-1 that recruits bone marrow-derived macrophage to the tumor area to promote vasculogenesis and tumor growth (Kioi et al. 2010). The response of tumor cells to radiation could also be regulated by autonomous pathways such as DNA damage repair, autophagy, reactive oxygen species (ROS) and mitochondrial respiration (Apel et al. 2008; Moding et al. 2013; Paglin et al. 2001). By comparing between intact or HIF-1α deleted in sarcoma cells, it has been shown that there was no significant difference in autonomous pathways after irradiation under normoxia (Zhang et al. 2015). However, further research is required to explore the precise mechanism of acquired radioresistant cells.
Next, we investigated the expression of the miR17-92 cluster in CRR cells and its regulatory effect on HIF-1α expression. Interestingly, we found that miR17-92 was down- regulated in CRR cells.
Leung et al. reported that exposure to fractionated radiation and single-dose radiation, down-regulated and up-regulated the miR 17-92 cluster in human breast cancer cells, respectively (Leung et al. 2014).
Taguchi et al. reported that miRNA 17-92 negatively regulates HIF-1α expression. They also showed that this negative regulatory effect is only exerted under normoxic conditions (Taguchi et al. 2008). We also found that miR 17-92 overexpression in CRR cells abolished the induction of HIF-1α following exposure to irradiation. These indicate that fractionated irradiation applied to establish CRR cells, might account for down regulation of miR17-92 and this, in turn, resulted in up regulation of HIF-1α in the CRR cells.
Adaptive responses might also explain accumulation of HIF-1α in CRR cells (Lall et al. 2014). It has been well-known that radiation through production of reactive oxygen species (ROS) can induce HIF-1α expression under normoxic conditions (Moeller et al. 2004). On the other hand, down-regulation of miR17-92 in CRR cells finely tunes the regulation of HIF-1α under normoxic conditions in favor of its stabilization.
Finally our result revealed that overexpression of miRNA-17-92 in CRR cells increased their radiosensitivity. However, in contrast with our findings, Jiang et al. reported that miR-17-92 overexpression by transfection in human mantle cell lymphoma cells markedly increased radioresistance (Jiang et al. 2010). In this study, we first established several CRR cell lines then overexpressed miR 17-92 in CRR cells. Supporting this notion, we also found that attenuated expression of miR19a resulted in the sensitization of the parental cells to irradiation and it’s up regulation conferred resistance to them. However, interestingly attenuated expression of miR 17-3p in parental cells resulted in refractory to irradiation suggesting down-regulation of this miR might be a key molecule to acquire the radioresistant phenotype. However further studies are required in this regard. Overexpression of miR 17-3p and miR19a by mimic in the CRR cells to evaluate the expression of HIF-1α after irradiation also would be subject to future study.
Supporting this notion, Xu et al. showed that miR-17-3p enhances the radiosensitivity of PC-3 cells by dysfunction of the three mitochondrial antioxidant enzymes. Furthermore, their study revealed that miR-17-3p increased ROS production and decreased mitochondrial respiration (Xu et al.2018).
In conclusion, we found that in CRR cells HIF-1α is accumulated under normoxic conditions in response to irradiation. However, HIF-1α is not causative of radioresistance. We also identified that miR17-92 cluster is down-regulated in CRR cells which, at least in part, accounts for HIF-1α overexpression. Interestingly, overexpression of miR17-92 in CRR cells canceled radiosensitivity in CRR cells. We also found that the downregulation of miR17-3p in the parental cells might be contributed to radioresistance. Our findings not only highlight the importance of miR17-92 in the acquisition of radioresistance but also introduce a new potential modality for cancer therapy. However further and comprehensive studies are required in this regard. For example, although we found that the expression of HIF-1 α and miR-17-92 cluster is inversely correlated in cancer cells tested it is imperative to show that HIF-1 α is a direct target of miR17-92 cluster, especially miR-17-3p or miR-19a. Luciferase assay would be a useful method to prove this.
References
Aebersold DM, Burri P, Beer KT, Laissue J, Djonov V, Greiner RH et al (2001) Expression of hypoxia-inducible factor-1α a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res 61:2911–2916
Apel A, Herr I, Schwarz H, Rodemann HP, Mayer A (2008) Blocked autophagy sensitizes resistant carcinoma cells to radiation therapy. Cancer Res 68:1485–1494
Arvold ND, Guha N, Wang D, Matli M, Deen DF, Warren RS et al (2005) Hypoxia-induced radioresistance is independent of hypoxia-inducible factor-1A in vitro. Int J Radiat Oncol Biol Phys. 62:207–212
Cellini F, Morganti AG, Genovesi D, Silvestris N, Valentini V (2014) Role of microRNA in response to ionizing radiations: evidences and potential impact on clinical practice for radiotherapy. Molecules 19:5379–5401
Dellas K, Bache M, Pigorsch SU, Taubert H, Kappler M, Holzapfel D et al (2008) Prognostic impact of HIF-1alpha expression in patients with definitive radiotherapy for cervical cancer. Strahlenther Onkol 184:169–174
Doi N, Ogawa R, Cui Z-G, Morii A, Watanabe A, Kanayama S et al (2015) The acquired radioresistance in HeLa cells under conditions mimicking hypoxia was attenuated by a decreased expression of HIF subunit genes induced by RNA interference. Exp Cell Res. 333:249–260
Feng Y, Liu J, Kang Y, He Y, Liang B, Yang P et al (2014) miR-19a acts as an oncogenic microRNA and is up-regulated in bladder cancer. J Exp Clin Cancer Res. 33:1
Fu Z, Chen D, Cheng H, Wang F (2015) Hypoxia-inducible factor-1α protects cervical carcinoma cells from apoptosis induced by radiation via modulation of vascular endothelial growth factor and p53 under hypoxia. Med Sci Monit: Int Med J Exp Clin Res. 21:318
Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466:835–840
Harada H (2011) How can we overcome tumor hypoxia in radiation therapy? J Radiat Res. 52:545–556
Harada H, Kizaka-Kondoh S, Hiraoka M (2005) Optical imaging of tumor hypoxia and evaluation of efficacy of a hypoxia-targeting drug in living animals. Mol Imaging. 4:182–193
Jiang P, Rao EY, Meng N, Zhao Y, Wang JJ (2010) MicroRNA-17-92 significantly enhances radioresistance in human mantle cell lymphoma cells. Radiat Oncol. 5:100
Kiani AA, Kazemi A, Halabian R, Mohammadipour M, Jahanian-Najafabadi A, Roudkenar MH (2013) HIF-1alpha confers resistance to induced stress in bone marrow-derived mesenchymal stem cells. Arch Med Res 44:185–193
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM (2010) Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Investig 120:694
Koukourakis MI, Giatromanolaki A, Sivridis E, Simopoulos C, Turley H, Talks K et al (2002) Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int J Radiat Oncol Biol Phys. 53:1192–1202
Kuwahara Y, Mori M, Oikawa T, Shimura T, Ohtake Y, Mori S et al (2010) The modified high-density survival assay is the useful tool to predict the effectiveness of fractionated radiation exposure. Int J Radiat Res. 51:297–302
Kuwahara Y, Oikawa T, Ochiai Y, Roudkenar M, Fukumoto M, Shimura T et al (2011) Enhancement of autophagy is a potential modality for tumors refractory to radiotherapy. Cell Death Dis. 2:e177
Kuwahara Y, Roudkenar MH, Urushihara Y, Saito Y, Tomita K, Roushandeh AM et al (2017) Clinically relevant radioresistant cell line: a simple model to understand cancer radioresistance. Med Mol Morphol. 50:195–204
Kuwahara Y, Tomita K, Urushihara Y, Sato T, Kurimasa A, Fukumoto M (2018) Association between radiation-induced cell death and clinically relevant radioresistance. Histochem Cell Biol 150:649–659
Ladelfa MF, Toledo MF, Laiseca JE, Monte M (2011) Interaction of p53 with tumor suppressive and oncogenic signaling pathways to control cellular reactive oxygen species production. Antioxid Redox Signal 15:1749–1761
Lall R, Ganapathy S, Yang M, Xiao S, Xu T, Su H et al (2014) Low-dose radiation exposure induces a HIF-1-mediated adaptive and protective metabolic response. Cell Death Differ 21:836–844
Leung C-M, Chen T-W, Li S-C, Ho M-R, Hu L-Y, Liu WS et al (2014) MicroRNA expression profiles in human breast cancer cells after multifraction and single-dose radiation treatment. Oncol Rep 31:2147–2156
Liu J, Zhang J, Wang X, Li Y, Chen Y, Li K et al (2010) HIF-1 and NDRG2 contribute to hypoxia-induced radioresistance of cervical cancer Hela cells. Exp Cell Res 316:1985–1993
Mendell JT (2008) miRiad roles for the miR-17-92 cluster in development and disease. Cell 133:217–222
Moding EJ, Kastan MB, Kirsch DG (2013) Strategies for optimizing the response of cancer and normal tissues to radiation. Nat Rev Drug Discov 12:526–542
Moeller BJ, Cao Y, Li CY, Dewhirst MW (2004) Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 5:429–441
Nordsmark M, Eriksen JG, Gebski V, Alsner J, Horsman MR, Overgaard J (2007) Differential risk assessments from five hypoxia specific assays: the basis for biologically adapted individualized radiotherapy in advanced head and neck cancer patients. Radiother Oncol 83:389–397
Paglin S, Hollister T, Delohery T, Hackett N, McMahill M, Sphicas E et al (2001) A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res 61:439–444
Patterson AV, Williams KJ, Cowen RL, Jaffar M, Telfer BA, Saunders M et al (2002) Oxygen-sensitive enzyme-prodrug gene therapy for the eradication of radiation-resistant solid tumours. Gene Ther 9:946–954
Poon E, Harris AL, Ashcroft M (2009) Targeting the hypoxia-inducible factor (HIF) pathway in cancer. Expert Rev Mol Med 11:e26
Ranasinghe WK, Baldwin GS, Bolton D, Shulkes A, Ischia J, Patel O (2014) HIF1α expression under normoxia in prostate cancer: which pathways to target? J Urol. 193:763–770
Schaue D, McBride WH (2015) Opportunities and challenges of radiotherapy for treating cancer. Nat Rev Clin Oncol. 30:120
Schoof CRG, da Silva Botelho EL, Izzotti A, dos Reis Vasques L (2012) MicroRNAs in cancer treatment and prognosis. Am J Cancer Res. 2:414
Semenza GL (2010) Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 29:625–634
Shimura T (2011) Acquired radioresistance of cancer and the AKT/GSK3β/cyclin D1 overexpression cycle. J Radiat Res. 52:539–544
Shimura T, Kakuda S, Ochiai Y, Nakagawa H, Kuwahara Y, Takai Y et al (2010) Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3beta-mediated cyclin D1 overexpression. Oncogene 29:4826–4837
Sprong D, Janssen HL, Vens C, Begg AC (2006) Resistance of hypoxic cells to ionizing radiation is influenced by homologous recombination status. Int J Radiat Oncol Biol Phys 64:562–572
Staab A, Fleischer M, Loeffler J, Said HM, Katzer A, Plathow C et al (2011) Small interfering RNA targeting HIF-1α reduces hypoxia-dependent transcription and radiosensitizes hypoxic HT 1080 human fibrosarcoma cells in vitro. Strahlenther Onkol 187:252–259
Taguchi A, Yanagisawa K, Tanaka M, Cao K, Matsuyama Y, Goto H et al (2008) Identification of hypoxia-inducible factor-1α as a novel target for miR-17-92 microRNA cluster. Cancer Res 68:5540–5545
Tomita K, Kuwahara Y, Takashi Y, Igarashi K, Nagasawa T, Nabika H et al (2018) Clinically relevant radioresistant cells exhibit resistance to H2O2 by decreasing internal H2O2 and lipid peroxidation. Tumour Biol. 40:1010428318799250
Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26:225–239
Vordermark D, Katzer A, Baier K, Kraft P, Flentje M (2004) Cell type–specific association of hypoxia-inducible factor-1α (HIF-1α) protein accumulation and radiobiologic tumor hypoxia. Int J Radiat Oncol Biol Phys. 58:1242–1250
Xu Z, Zhang Y, Ding J, Hu W, Tan Ch, Wang M et al (2018) miR-17-3p downregulates mitochondrial antioxidant enzymes and enhances the radiosensitivity of prostate cancer cells. Mol Ther Nucleic Acids. 13:64–77
Zhou S, Ye W, Ren J, Shao Q, Qi Y, Liang J et al (2015) MicroRNA-381 increases radiosensitivity in esophageal squamous cell carcinoma. Am J Cancer Res. 5:267
Ye W, Qin F, Zhang J, Luo R, Chen H-F (2012) Atomistic mechanism of microRNA translation upregulation via molecular dynamics simulations. PLoS ONE 7:e43788
Zhang M, Qiu Q, Li Z, Sachdeva M, Min H, Cardona DM et al (2015) HIF-1 alpha regulates the response of primary sarcomas to radiation therapy through a cell autonomous mechanism. Radiat Res. 183:594–609
Zhao L, Lu X, Cao Y (2013) MicroRNA and signal transduction pathways in tumor radiation response. Cell Signal 25:1625–1634
Funding
We acknowledge department of Pathology, Institute of Development, Aging and Cancer, Tohoku University, Sendai, Japan who hosted the research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Roudkenar, M.H., Fukumoto, M., Roushandeh, A.M. et al. Disturbance in the regulation of miR 17-92 cluster on HIF-1-α expression contributes to clinically relevant radioresistant cells: an in vitro study. Cytotechnology 72, 141–153 (2020). https://doi.org/10.1007/s10616-019-00364-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-019-00364-9