
Abstract
This study aimed to develop a bovine mammary epithelial (BME) cell line model, which provides a possibility to determine functional properties of the bovine mammary gland. The primary cell culture was derived from bovine mammary gland tissues and processed enzymatically to obtain cell colonies with epithelial-like morphology. The cultures of BME cells were purified and optimally cultured at 37 °C in DMEM/F12 medium supplemented with 10% fetal bovine serum. The BME cells were identified as epithelial cell line by the evaluating the expression of keratin-18 using immunofluorescence staining. A novel gene expression system strongly enhances the expression of telomerase, has been used to immortalize BME cell line termed hTBME cell line. Interestingly, telomerase remained active even after over 60 passages of hTBME cell line, required for immortalization of BME cells. In addition, the hTBME cell line was continuously subcultured with a spontaneous epithelial-like morphology, with a great proliferation activity, and without evidence of apoptotic and necrotic effects. Further characterization showed that hTBME cell line can be continuously propagated in culture with constant chromosomal features and without tumorigenic properties. Finally, established hTBME cell line was evaluated for mammary gland specific functions. Our results demonstrated that the hTBME cell line was able to retain functional-morphological structure, and functional differentiation by expression of beta (β)-casein as in the bovine mammary gland in vivo. Taken together, our findings suggest that the established hTBME cell line can serve as a valuable tool for the study of bovine mammary gland functions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The bovine mammary gland is a cutaneous mammary-like sweat gland, which is composed of secretory tissue and ductular system supported by the connective tissue. Functional differentiation of the mammary gland occurs with apparent morphological changes of the epithelial cells and allows milk production and secretion (Richert et al. 2000). The alveolar epithelial cells are the functional unit of the mammary gland that synthesize and secrete milk (Anand et al. 2012; Capuco et al. 2002). Production of milk by mammary gland starts during pregnancy and is characterized by expression of essential genes for milk synthesis, such as casein and lactalbumin (Neville et al. 2002). Expression of milk protein genes in epithelial cells of bovine mammary gland depends on different factors. At parturition stage, hormonal changes take place to prepare mammary gland for secretion of colostrum and then milk (Barber et al. 1992). After parturition with few days, growth and proliferation of the mammary gland cells start to be increased. While during the lactation period, there is a little growth, and a slow proliferation rate of the mammary gland cells (Capuco et al. 2002).
Understanding of the milk production process has important implications for consumers, producers, and animals concerned by production efficiency of the milk and animal health care (Kozłowski and Motyl 2007; Povey and Osborne 1969). Considerable efforts have been devoted to better understanding the mechanism that is involved in regulating the bovine mammary gland development in order to generate information that may be useful in (1) increase efficiency of milk production, and (2) reduce response of mammary gland to pathogenic infections (Jedrzejczak 2010; Kozłowski and Motyl 2007). However, the mechanism of milk production and cellular complexity of bovine mammary gland makes it complicated to study the functional role of each different components of the mammary gland in vivo. In addition, the study of biological properties of bovine mammary gland tissue in vitro requires some approaches that cannot be achieved without the use of the suitable cell culture system. Since BME cells represent a valuable in vitro model for development and tissue engineering, therefore, a variety of mammary primary culture systems has been developed to facilitate studies on the milk production process and the induction of milk protein gene expression. However, primary culture system has major drawbacks such as the short lifespan of cells. Therefore, several immortalized mammary gland epithelial cell lines have been established either with (1) transfection of the normal mammary epithelial cell with the simian virus 40 Large T (SV40LT) gene (Huynh et al. 1991; Serakinci et al. 2007; Zavizion et al. 1995b, 1996) or (2) transfection of cells with telomerase reverse transcriptase (TERT) gene (Gao et al. 2008; Kim et al. 2002; Toouli et al. 2002; Zhao et al. 2010a). However, some of these immortalized cell lines maintained on their proliferation for prolonged time periods without maintaining their specific functions like the natural cells of the mammary gland. Therefore, the goal of this study was to establish a new immortalized cell line from bovine mammary gland tissue, which can offer cells more likely to represent in vivo condition of the mammary gland. The human TERT (hTERT) has been used in immortalization of different cell lines, for example, Lee et al. (2004) have shown that transfer of an exogenous hTERT, encoding the catalytic subunit of human telomerase, can be used to overcome telomere controlled-senescence by preventing shortening telomere. In contrast, it was also reported that many hTERT positive cell colonies could not escape from irreversible cellular senescence (Zhao et al. 2010a). However, recent reports have been suggested that telomerase activity is required for cellular immortalization in pig cells depending on culture conditions (Christman et al. 2006; Davis and Kipling 2005; Wong et al. 2003).
It was of great significance in this study to extend the life span and to retain spontaneous immortalization of mammary epithelial cell in culture. To achieve such purpose, a novel gene expression system using pCI-neomycin (neo) plasmid (kind gift from Wang Li, School of Stomatology, Fourth Military Medical University) was used. The hTERT cDNA flanking chimeric intron was inserted into the pCI-neo plasmid upstream of the CMV immediate-early enhancer/promoter. The hTERT cDNA was inserted upstream of the chimeric intron in order to prohibit utilization of possible cryptic donor splice sites within cDNA sequence (Senapathy et al. 1990). The SV40 late polyadenylation (polyA) signal is positioned downstream from hTERT. Neomycin selectable marker is presented in pCI upstream of the SV40 early enhancer/promoter. Moreover, further analyses were carried out in this study to clarify functional and morphological properties of the newly established hTBME cell line. The results obtained from this study can be useful in many studies related to function and structure of bovine mammary gland.
Materials and methods
Tissue collection and culture condition of BME
Normal mammary gland tissue was obtained from a local slaughterhouse (Shannxi, Xian, China) according to a protocol approved by Institutional Animal Care and Use Committee. The mammary glands were dissociated mechanically and then digested in serum-free media containing 0.2% collagenase II solution (C6885, Sigma, St. Louis, MO, USA) for 90 min. The digested cells were sequentially sieved through a 100 and 40 µm cell strainer (Falcon, Corning) to obtain single-cell suspensions. The cell suspension was washed three times using fresh phosphate buffer saline (PBS) and then transferred to tissue culture dishes (CLS430166, Corning, Corning, NY, USA) for 1 h to enable adherence of fibroblasts at 37 °C in DMEM/F12 medium supplemented with 10% fetal bovine serum (FBS; 26140079, Gibco, Grand Island, NY, USA) and 1% penicillin–streptomycin in a humidified atmosphere with 5% CO2. The unattached cells were converted to a new petri dish and incubated at 37 °C and humidified atmosphere with 5% CO2 in DMEM/F12 medium supplemented with 10% FBS and 1% penicillin–streptomycin and 10 ng/mL epidermal growth factor (EGF; E9644, Sigma) for 7 days. The digestion step was repeated until purified epithelial cells were obtained.
Immunofluorescence (IF) assay
For identification of purified BME and hTBME cell lines as an epithelial cell line, IF assay was used exactly as previously described (Said et al. 2014). Briefly, BME, hTBME and bovine fibroblast cells as a negative control were fixed with 4% paraformaldehyde in PBS for 15 min at RT and washed with PBS 3 times. The cells were followed by permeabilization with 0.1% Triton X-100 for 20 min. After three times of washing with PBS, free binding sites were blocked using PBS containing 0.1% bovine serum albumin (BSA; A9418, Sigma) for 1 h at RT. Cells were incubated with mouse monoclonal anti-keratin 18 antibody (CK18; MAB3234, Chemicon, Temecula, CA, USA) for overnight at 4 °C. After washing with PBS, cells were incubated with secondary antibody (CY3-goat anti-mouse IgG (A10521, Invitrogen, Carlsbad, CA, USA) at 1:500 dilution for 30 min at RT. Nuclear staining was performed using Hoechst 33342 (H3570, ThermoFisher, Waltham, MA, USA) for 10 min in the dark. The cells were inspected under a Nikon Inverted Microscope Eclipse Ti-S. To detect cellular proliferation of the BME cell line at passage 15 and hTBME cell line at passage 60, IF assay was used exactly as mentioned above. Rabbit anti-human Ki67 antibody (PA1918, Boster) and CY3-goat anti-rabbit IgG (BA1032, Boster, Pleasanton, CA, USA) were used as primary and secondary antibodies, respectively. Nuclear staining was done using Hoechst 33342. The cells were observed under Nikon Inverted Microscope Eclipse Ti-S. For quantitative analysis of proliferation marker, Ki67 expression in proliferating cell line, data were reported as percentages of labeled cells over the total number of cells. The value represents the average of two independent experiments.
Transfection and cell selection
Transfection was performed as previously described (Said and Osterrieder 2014; Wang et al. 2015). The purified BME cell line was transfected with pCI-neo_hTERT using lipofectamine® 2000 Reagent (11668027, Invitrogen). After 3 weeks selection with 300 µg/mL G418 (A1720, Sigma), each cell clone was seeded in culture plates of 96 wells for further selection of resistant cells, which expressed hTERT. The BME clone expressing hTERT (hTBME) was cultured in DMEM/F12 with 10% FBS.
Quantitative telomerase detection (QTD) real-time PCR assay
To determine telomerase activity of hTBME cells, BME cells at passage 5 and hTBME cells at passage 25, 45 and 60 were used. Telomerase activity was detected as described previously (Hou et al. 2001) with some modifications. The cell extracts for telomerase activity assays were prepared following manufacturer’s instructions of quantitative telomerase detection kit (MT3010, Allied Biotech Inc., Ijamsville, MD). The protein concentration in each extract was measured using the BCA protein Assay Reagent Kit (23225, Pierce, Rockford, IL, USA). Real-time polymerase chain reaction (PCR) was performed using 12.5 µL premix, 1.0 µL cell extracts, and 11.5 µL water. The PCR condition consisted of two cycles of 20 min at 25 °C and 10 min at 95 °C followed by 40 three-step cycles of 30 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C. The analysis of PCR results was performed using ANOVA and the Fisher PLSD multiple comparisons test.
Growth studies and apoptosis assay
To determine growth properties of transfected hTBME cells, untransfected BME cells at passage 15 and hTBME cells at passage 25, 45 and 60 were cultured at a density of 1 × 104 cells per well of 24-wells plate. Growth curves were determined from a daily count of cells using countess™ automated cell counter (C10227, Invitrogen) from two replicate cultures stained with trypan blue (T10282, Invitrogen) The experiment was performed in three independent replicates and each sample was also tested in duplicate for each experiment.
Flow cytometry was used to detect cell cycle of BME and hTBME cells. The BME cells of passage 5 and hTBME cells of passage 60 were trypsinized and suspended in fluorescent-activated cell sorter (FACS) buffer (PBS containing 2% FBS and 0.01% sodium azide. The cells were washed three times with FACS buffer and stained with propidium iodide (PI) with a final concentration of 25 mg/mL at 4 °C for 30 min. The cells were detected with an FACSCalibur flow cytometer (BD FACSAria™ II), and the intensity of fluorescence was analyzed using BD CellQuest™ Pro software. The experiment was performed at three independent times.
For detection of cell apoptosis, Annexin V-FITC apoptosis detection kit (Beyotime Institute of Biotechnology, C1017, Shanghai, China) was used as described by manufacturer’s instruction. The BME cells at passage 5 and hTBME cells at passage 60 were collected by centrifugation at 1000 r/min for 3 min and washed twice with cold PBS. The cells were gently resuspended in Annexin V binding buffer, and then incubated with Annexin V-FITC/PI for 15 min in the dark. The apoptotic cells were detected with an FACSCalibur flow cytometer (BD FACSAria™ II) and analyzed using BD CellQuestTM Pro software.
Karyotype analysis and soft agar assay
Metaphase preparations were performed using a procedure reported previously (Gao et al. 2008; Moore et al. 1999) with some modifications. The hTBME cells were treated with 0.5 µg/mL colcemid (D7385, Sigma) for 6 h, and then trypsinized and collected by centrifugation at 1000 rpm for 10 min. The cells were then hypotonically treated with 0.075 M KC1 for 30 min at 37 °C and fixed with methanol-acetic acid solution (3:1) for 10 min. Cells suspensions were spread on glass slides and then stained with Giemsa solution for 10 min. A total of 20 randomly selected metaphase spreads were analyzed with a Nikon Microphot-FX microscopy.
Soft agar assay was performed exactly as described before (Anand et al. 2012; Wang et al. 2015). Briefly, each well of 6-well culture plates was first coated with 0.6 % agarose gel (A9045, Sigma) in the bottom layer. A 0.3% agarose gel mixture containing either BME cells at passage 5, hTBME cells at passage 60 or HeLa cells as a positive control were layered on top of the 0.6 % agarose gel and then incubated at 37 °C in a humidified atmosphere with 5% CO2 for 3 weeks. Colonies were inspected under an OLYMPUS microscope (CKX41, Olympus Life sciences, Tokyo, Japan). The colonies were contained more than ten viable cells, were scored positive.
Transmission electron microscopy (TEM)
In preparation for TEM, hTBME cells at passage 60 were digested with 0.25% trypsin and 0.08% EDTA after having been washed three times with PBS. Then cells were transferred to 10 mL centrifuge tubes and washed with cold PBS (0.1 M, pH = 7.4). The cell precipitation was packaged with 1% agarose gel, cut into small pieces about 1 mm thick, and fixed by osmic acid for 2 h. The cells were then rapidly dehydrated with a serial dilution of ethanol solution (i.e., 50, 70, 80, 90, 95 and 100%). Each specimen was immersed in acetone–epoxy resin solution and then embedded in Epon-812 (45345-250ML-F, Sigma) for 24 h. Ultrathin sections were stained using uranyl acetate and counterstained with alkaline lead citrate. The ultrastructure of the hTBME cells were observed under a transmission electron microscope (JEM-2000EX, JEOL Ltd., Tokyo, Japan).
RT-PCR and western blot anaysis
For analysis of casein expression in established hTBME cell line, RT-PCR and Western blot were performed using a procedure described previously (Anand et al. 2012) with minor modifications. The hTBME cell line at passage 60 was cultured for 48 h at 37 °C and 5% CO2 in DMEM/F12 supplemented with 10% FCS, 10 ng/mL epidermal growth factor (E9644, Sigma), 1 µg/mL hydrocortisone (H 4001, Sigma), 5 µg/mL insulin (11507, Sigma), and 5 μg/mL prolactin (L6520, Sigma). Cells were collected to investigate the secretion ability of hTBME cells. For RT-PCR, total cellular RNA from established hTBME cell line, bovine mammary tissue as a positive control and bovine fibroblast as a negative control were prepared using TRIzol® LS Reagent (10296028, ThermoFisher). Possible genomic DNA contamination in RNA preparation was removed by using DNA-free kit (AM1906, Invitrogen). The design of specific oligonucleotide primers for GAPDH was as follows: forward primer 5′-ACGGCACAGTCAAGGCAGA-3′ and reverse primer 5′-GTGATGGCGTGGACAGTGG-3′ and for β-casein were as follows: forward primer 5′-AGGAACAGCAGCAAACAG-3′ and reverse primer 5′-TTTCCAGTCGCAGTCAAT-3′. The RNA was reverse transcribed into cDNA using SuperScript™ II Reverse Transcriptase (18064014, Invitrogen) according to the manufacturer’s protocol. The PCR reaction was performed using the following PCR condition: 95 °C for 3 min, 95 °C for 30 s, 58 °C for 30 s, 72 °C for 1 min for 30 cycles and 72 °C for 5 min in the last cycle.
For Western blot analysis, established hTBME cell line, and bovine mammary tissue as a positive control and bovine fibroblast as a negative control were used. Pellets of cells were resuspended in radioimmunoprecipitation assay (RIPA) buffer (1 mM Tris, pH 7.4, 1% Triton X-100, 0.25% sodium deoxycholate, 5 M sodium chloride, 0.5 mM EDTA, 0.1% sodium dodecyl sulfate [SDS]) and a protease inhibitor cocktail (04693116001, Roche, Mannheim, Germany). Samples were mixed with sample loading buffer (1 M Tris–HCl, pH 6.8, 0.8% SDS, 0.4% glycerol, 0.15% β-mercaptoethanol, 0.004% bromophenol blue), and heated in boiling water for 10 min. The samples were separated on 10% polyacrylamide gels and transferred to PVDF membrane using semi-dry transblot apparatus (#10600021, GE Healthcare, Shanghai, China). The membrane was incubated with primary antibody of mouse anti-β-casein monoclonal antibody (MAb) and mouse anti-GAPDH MAb (G8795, Sigma) as a loading control. The horseradish peroxidase-conjugated goat anti-mouse was used as a secondary antibody. Reactive bands were visualized by enhanced chemiluminescence (Sc-2048, ECL plus, ThermoFisher).
Results
Isolation of BME and establishment of immortalized hTBME cell lines
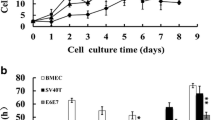
A heterogeneous population of epithelial and fibroblast-like cells was obtained by collagenase type II digestion of bovine mammary gland tissue. Further passages with selective collagenase resulted in the removal of spindle-shaped fibroblasts and gradually yielded a homogenous population of cuboidal-shaped epithelial cells. After 5 passages, fibroblast cells died and number of epithelial cells gradually increased, and exhibited a typical epithelial-like morphology on reaching confluency (Fig. 1a). To detect whether the epithelial marker, Keratin 18 can be expressed in isolated purified BME cell line, IF assay was performed using mouse monoclonal antibody that targeting the epithelial marker, keratin 18. Our results demonstrated that isolated BME cell line was reactive with keratin 18-specific antibody but not with the bovine fibroblast cell line (Fig. 1b). Thereafter, isolated purified BME cell line was used for exogenous transfection with pCI-neo_hTERT, encoding catalytic subunit of telomerase. The transfected cells that expressed hTERT were selected after 3 weeks using G418. In order to purify a resistant cell line, cells were digested, diluted and one positive clone, named hTBME clone was obtained. After a period of culture, the hTBME clone gradually increased in size, and exhibited a typical tightly packed and cobblestone-like morphology on reaching confluency. After 60 passages, the established cell line had a morphology similar to the early passage of BME cell line (Fig. 1a). In addition, expression of keratin 18 in the established hTBME cell line was detected (Fig. 1b). Taken together, we concluded from our findings that the immortalized hTBME cells were successfully established and retained their epithelial-like morphology even after over 60 passages.

Morphology and immunofluorescence detection of keratin 18 expression in BME and immortalized hTBME cell lines. a Formation of a typical epithelial-like morphology on reaching confluency after serial passages of BME cell line (left). Immortalized hTBME cell at passage 60 showed a typical cobblestone pattern of the freshly isolated primary BME cell line (right). Magnification: ×20, scale bar = 20 µm. b For immunofluorescence, immortalized hTBME at passage 60, purified BME and bovine fibroblast cell line as a negative control were stained for the epithelial marker, keratin-18. CY3-goat anti-mouse IgG was used as a secondary antibody. Hoechst 33342 was used as a nuclear counterstain. Magnification: ×10, scale bar = 100 µm. Results represent images from three independent experiments
Extension of telomerase activity in the established hTBME cell line
In order to quantify the extent of telomerase activity either in the BME cell line at passage 5 or in the established hTBME cell line at passage 25, 45, and 60, quantitative telomerase detection (QTD) Real-Time PCR Assay was performed. A standard curve was generated using the reading of the threshold (CT) of Real-Time PCR. The results showed that telomerase activity during the 5th passage of the BME cell line was a significantly lower in comparison with telomerase activity detected in the hTBME cell line (Fig. 2). In contrast, the hTBME cell line at different times of the passage demonstrated a dramatic increase in telomerase activity, which remained active until the 60th passage of the cells with significantly higher level than for the early passage of the BME cell line at passage 5 (Fig. 2). Our findings indicated that telomerase activity level is insufficient for immortalization and continuous passage of non-transfected BME cell line. While, detectable telomerase level in all passages of the established hTBME cell line was sufficient for spontaneous immortalization of hTBME cells in culture.

Detection of telomerase activity. Quantitative telomerase detection (QTD) real-time PCR assay was used to detect telomerase activity in the BME cell line at P5 and the established hTBME cell line at P25, 45 and 60. Errors bars represent standard deviations from three independent experiments. The asterisk denotes statistically significant differences between different passages of the cell lines (p < 0.05)
Growth and proliferation characteristics of the hTBME cell line
To determine the growth rate record of the established hTBME cell line, growth kinetics were performed out either for the BME cell line at passage 15 or the hTBME cell line at passage 25, 45 and 60. Growth rate capability of the hTBME cell line showed a rapid growth rate at 3–6 day and reached a maximal growth rate nearly at the 7th day of culture. Then, the number of cells started to be reduced by the 8th day of culture. While the BME cell line showed a very limited degree of proliferation in comparison with hTBME cell line (Fig. 3a). Population doubling time (PDT) of hTBME was calculated according to the previous studies (Hong et al. 2007; Wang et al. 2015). Our results revealed that the PDT of hTBME was approximately 54 h without any distinct changes between different passages of the established cell line (Fig. 3a).

Comparison between growth and proliferation characteristics of the BME and hTBME cell lines. a Cell growth rates of the BME cell line at P15 and the hTBME cell line at P25, 45 and 60. Cells were collected at different time points (0, 1, 2, 3, 4, 5, 6, 7 and 8 days). Data are represented as mean ± SD of three independent experiments. b Analysis of cell cycle of the hTBME cell line at P60 (left) and the BME cell line at P5 (right) by flow cytometry. The PI fluorescent dye was used to stain cellular DNA. The fluorescence intensity of cells was measured in the G1, S and G2 phases. (c) and (d) Proliferation analysis during differentiation of the BME cell line at P5 and the hTBME cell line at P60. c Immunofluorescence assay was used to detect expression of proliferation marker, Ki67 in the BME and hTBME cell lines. Magnification: ×20, scale bar = 20 µm. d Quantitative analysis of proliferation marker expression during differentiation of the BME and hTBME cell lines. The number of total cells and labeled cells was counted in five randomly chosen fields from duplicate experiments, after Ki67 immunostaining. Data are expressed as percentages of Ki67+ cells of the total number of cells. Data are shown as mean ± SD of two independent measurements. The asterisk denotes statistically significant differences (p < 0.05)
Further analysis of cell cycle of the established hTBME cell line was determined using flow cytometry. The BME cell line of passage 10 and hTBME cell line of passage 60 were treated with a fluorescent dye, PI that stains cellular DNA. Amount of DNA in each cell line was correlated to the fluorescence intensity of stained cells at certain wavelengths. As shown in Fig. 3b, fluorescence intensity of the non-transfected BME cells in Gap 1 (G1), synthesis (S) and Gap 2 (G2) phases were 72.55, 25.70 and 1.75%, respectively, while in the transfected hTBME cells were 55.45, 42.80 and 1.75%, respectively. Our results demonstrated that growth arrest in the G1 phase was higher in BME cells than hTBME cells. While the proportion of cells in the S phase was increased in hTBME cells compared to BME cells, suggesting that the immortalized cell line has a higher proliferative capacity than non-transfected cell line. Furthermore, proliferation marker Ki67 was detected either in BME cells at passage 15 and hTBME at passage 60 using IF assay. Our findings demonstrated that most of the hTBME cells were Ki67 positive. In contrast, a majority of MEC cells were not stained by anti-Ki67 antibody except very few cells, which stained lightly (Fig. 3c). Quantitative immunofluorescence analysis revealed that 17 and 89% of BME and hTBME cells, respectively, were positive (Fig. 3d). Overall, we conclude that the established hTBME cells in this study are characterized by a fast growth rate and a great proliferative activity.
Chromosomal and tumorigenic transformation analysis of established hTBME cell line
In order to address chromosomal structure of the established hTBME cells, chromosomal analysis of hTBME cells at the 60th passage was carried out by randomly selecting the field of vision. Our results demonstrated that hTBME cells had a normal diploid configuration containing 60 chromosomes (Fig. 4a), which are specific for bovine origin cells (German and Barash 2002; Hu et al. 2009). The chromosomal analysis of the established cell line was performed twice. In order to determine whether the hTBME cell line has the ability to transform from normal to tumorigenic cell line in vitro, a soft agar experiment was performed. Our results showed that the HeLa cell as a positive control was able to form colonies on soft agar as previously reported (Kwak et al. 2006), but our established hTBME cell line was not able to show any colonies on agar (Fig. 4b). Taken together, we concluded from our results that the hTBME cell line can be reproduced without (1) a chromosomal abnormality during its division, and (2) a tumorigenic transformation.

Chromosomal and tumorigenic potential analysis of the established hTBME cell line. a Chromosomal analysis of the established hTBME cell line. Representative metaphase spread and karyotype of the hTBME cell line showing 30 pairs of chromosomes specific to bovine origin cells (2n = 60). The chromosomal analysis was performed twice. b In vitro tumorigenicity study of the hTBME cell line. Colonies formation either for hTBME (left) or Hela (right) cell lines were imaged using an inverted microscope (OLYMPUS CKX41). Magnification: ×20, scale bar = 20 µm
Escape of the established hTBME cell line from apoptotic and necrotic effects
Furthermore, to examine whether primary culture BME and established hTBME cell lines escape from apoptosis, flow cytometry assay was used. The BME cells at the early stage of passages and hTBME cells at passage 60 were stained with PI/AnnexinV–FITC. From our results, we observed that the BME cell line stained positive either for PI and AnnexinV–FITC with 18.5 and 7.4%, respectively, from the total number of the cells. While the hTBME cell line stained positive for PI with 5.6 and 3% for AnnexinV–FITC (Fig. 5). From our findings, we concluded that the total number of BME cells, which stained with PI/AnnexinV–FITC was significantly increased compared with the total number of hTBME cells. Taken together, our findings suggest that the hTBME cells can normally be grown without an evidence of any change in their growth properties even after the 60th passage cell.

Analysis of apoptotic and necrotic effects in hTBME and BME cells. The established hTBME cell line at passage 60 (right) and BME cell line at passage 5 (left) were stained with annexin V and propidium iodide (PI) and analyzed by flow cytometry. Four populations of cells were identified: B1 [necrotic (PI) only], B2 [apoptotic/necrotic (annexin V + PI)], B3 [live (no stain)], and B4 [apoptotic (annexin V only)]
Ultrastructural characterization of the established hTBME cell line
In order to address ultrastructure of the established hTBME cell line of passage 60, TEM was used. The results of TEM showed that our established cell line was regularly arranged in net rows, oval, and/or round shape and vacuolated nuclear envelope were visible (Fig. 6a). The surface of the cell was characterized by a high microvilli density that increases cell surface area and is involved in wide variety of functions, for example, secretion, absorption and cellular adhesion (Fig. 6b). The cytoplasm possessed a considerable quantity of secretory vesicles and lipid droplets, indicated strong secretory activity of established cell line (Fig. 6c). The endoplasmic reticulum was prominent in the established cells (Fig. 6d). These findings suggest that our established cell line tends to maintain its morphological and metabolic state, implying that the hTBME cell line can be reproduced with a normal morphological shape similar to natural mammary gland cells.

Ultrastructural analysis of the hTBME cell line at passage 60 using TEM. a Electron microscopy of the hTBME cell line showing a cuboidal appearance. Morphological analysis of the hTBME cell line revealed the presence of abundant microvilli (MV), scale bar = 2 µm (b), secretory vesicles (SV) and lipid droplets (LD), scale bar = 1 µm (c), and endoplasmic reticulum (ER), scale bar = 0.5 µm (d)
Milk protein expression and dome formation in the established hTBME cell line
To evaluate hormonal responsiveness in the established hTBME cells at passage 60, β-casein expression was measured by RT-PCR and Western blot. For RT-PCR assay, expression of β-casein at mRNA level in the established hTBME cells, bovine mammary tissue and bovine fibroblast cells as a positive and a negative control, respectively, was measured. The housekeeping gene Glyceraldehyde 3–phosphate dehydrogenase (GAPDH) was used as a loading control. Our results demonstrated a robust amplification of the β-casein in bovine mammary tissue cells and hTBME cells in presence of insulin, hydrocortisone, and prolactin but not in bovine fibroblast cells (Fig. 7a, left panel). Furthermore, Western blot assay was conducted to determine casein secretion in cultured hTBME cells in presence of insulin, hydrocortisone and prolactin using anti- β-casein antibody. Western blot results revealed that lysate of hTBME and bovine mammary tissue reacted positively with anti- β-casein antibody while no reactivity was observed in the lysate of fibroblast cells, which were used as a negative control (Fig. 7a, right panel). We concluded from our results that hTBME cells are able to induce expression of β-casein at mRNA and protein levels.

Analysis of β-Casein expression and dome formation in the established hTBME cell line. a RT-PCR and western blot were used to detect β-Casein expression. For RT-PCR (left) and western blot (right), hTBME at passage 60, bovine mammary tissues as a positive control and bovine fibroblast cells as a negative control were used. Loading control represents the GAPDH and β-actin in RT-PCR and western blot, respectively. Results are representative of three independent experiments. b Phase contrast image of dome structure in the hTBME cell line treated with hydrocortisone, 5 µg/mL insulin, and prolactin (right) and the non-treated hTBME cell line (left). Magnification: ×20, scale bar = 20 µm
The phase contrast microscope was used to visualize domes formation in the established hTBME cell line either in the absence or presence of insulin, hydrocortisone and prolactin (Fig. 7b).
Overall, these data together suggest that our established hTBME cell line experimentally mimic mammary gland development at the cellular level.
Discussion
Although a variety of in vitro models of mammary culture systems have been developed, there is still difficulty in establishing a suitable in vitro model system, which more accurately mimics the bovine mammary gland in vivo. Therefore, this study aimed to establish a spontaneous immortalized bovine mammary epithelial cell line and to characterize its functional properties in culture. Collagenase enzyme was used to digest primary cell culture, which was derived from bovine mammary gland tissue (Anand et al. 2012; Wang et al. 2015). The enzymatic digestion of cells yielded a heterogeneous population of cells containing epithelial and fibroblast-like cells. Fibroblast cell lines were removed from epithelial cell lines by digestion of the extracellular matrix that allows a more efficient disaggregation of epithelial cells (Booth and O’Shea 2002). The existence of cell colonies with epithelial-like morphology, named bovine mammary epithelial (BME) cells, were purified and optimally maintained at 37 °C in DMEM/F12 medium supplemented with 10% FBS. BME cells exhibited a cobblestone-like morphology on reaching confluency similar to characteristic features of mammary epithelial cells of other species, including bovine (Ahn et al. 1995; Gibson et al. 1991; Huynh et al. 1991; Shi et al. 2014; Talhouk et al. 1990; Tong et al. 2012), and formed islands of cells when spread at low density. Thereafter, the isolated purified BME cell line differentiated into a more mature cell, and successfully frozen and stored in liquid nitrogen. The ability of cells to functionally recover from cryopreservation was confirmed. The epithelial characteristic of purified BME cells was investigated using immunofluorescence assay. Cytokeratins are intermediate filament proteins found in most epithelial cells and have been used as markers of mammary epithelial cells (Fuchs 1988; Iannuzzi 1994). It was reported that keratin-18 has been used to determine epithelial cell property (Fu et al. 2014; Schmid et al. 1983; Taylor-Papadimitriou et al. 1989). Thus, in our study, keratin-18 expression was used to provide an evidence of epithelial nature of the purified BME cells.
Immortal cell populations from bovine mammary epithelial cells have been previously generated. However, at terminal differentiation of the immortalized cells, senescence was accompanied by irreversible changes in both morphology and gene expression (Kim et al. 2002; Toouli et al. 2002; Zhao et al. 2010a). Recently, some studies have used telomerase in establishment of immortalized cell line to control senescence of the cells (Bodnar et al. 1998; Jiang et al. 1999; Ouellette et al. 2000; Vaziri and Benchimol 1998), and to avoid causing cancer-associated changes or altering phenotypic properties (Jiang et al. 1999; Morales et al. 1999; Ouellette et al. 2000). In this study, the pCI-neo vector expressing hTERT was used to establish a new immortalized cell line, termed hTBME cell line. Strong expression of hTERT was achieved under the CMV immediate-early enhancer/promoter. The pCI-neo also contains a chimeric intron downstream of the CMV promoter and upstream of the hTERT. This intron is important to enhance expression of all inserted cDNA at high level (Brinster et al. 1988). The SV40 late polyA signal was used to terminate transcription of the inserted hTERT gene. It has previously been reported that the SV40 late polyA signal is considerably efficient to enhance stability and translation of RNA approximately fivefold more than the SV40 early polyA signal (Bernstein et al. 1989). Interestingly, our findings showed that established hTBME cell line was spontaneously immortalized with a typical epithelial-like morphology even after passage of cells over more than 60 times. Our findings match results of previous studies, which also showed that hTERT was used to immortalize normal human cells to maintain their differentiation potential without any morphological changes (Herbert et al. 2002; Jiang et al. 1999; Lee et al. 2004; Morales et al. 1999, 2003; Ouellette et al. 2000; Ramirez et al. 2003; Yang et al. 1999; Yudoh et al. 2001). However, it has been previously shown that some of the established cell lines could gain immortalization, but after several times of passages those cell lines lost immortalization and became senescent (Zhao et al. 2010a). In order to gain insight into whether immortalization in our established hTBME cell line was constant even after several passages, different experiments were done in this study. Our findings proved that telomerase was active in the immortalized hTBME cell line even after 60 passages of the cells. Telomeres are important structures that cap and protect the ends of linear chromosomes. In addition, it has been previously known that reduction in telomerase activity leads to cessation of cell cycle phases (Lee et al. 2004). To correlate our results with the previous report, interestingly, we observed that cell cycle phases of immortalized hTBME cell line were active, and clearly under a distinct proliferation capability. Therefore, we have successfully maintained the hTBME cell line in a culture for more than 60 passages with evidence of immortalization gain.
It is essential to record growth properties of a newly immortalized cell line before starting any experiment because any changes in growth characteristics of cells can result in a significant effect on results of further experiments (Geraghty et al. 2014). Growth curve analysis of the established hTBME cell line demonstrated a typical growth pattern, and a population doubling time of about 54 h, without any significant changes between different passages of hTBME cell line. Similar observations have been reported (Anand et al. 2012; Hu et al. 2009; Zavizion et al. 1995a). The cell cycle analysis using flow cytometry showed that the immortalized cell line had a proliferative advantage and a distinct proliferation capability. Moreover, quantitative immunofluorescence analysis was performed using the proliferation marker Ki67 to prove that the hTBME cells cell line had a high proliferative activity in agreement with a previous report (German and Barash 2002). Our conclusion from these data is that the established hTBME cell line can be continuously grown with a fast growth rate and a great proliferative activity even after more than 60 passages of the cells. In addition, the established hTBME cell line exhibited a chromosomal number similar to earlier reports in bovine (German and Barash 2002; Hu et al. 2009), implying that the immortalized cell line proliferated normally without any abnormalities in its chromosomal number. Inactivation of tumor suppressor proteins and activation of oncoproteins result in transformation of cells (Hanahan and Weinberg 2000). It was essential to address whether the established hTBME cell line has transforming properties, interestingly, the immortalized hTBME cell line showed normal growth characteristics and no tendency to malignant transformation. This observation was similar with a previous study (Zhao et al. 2010a). Subsequently, the established hTBME cell line can be used to study factors, which are involved in mammary gland carcinogenesis.
Apoptosis can be induced in mammary gland cells after removal of lactogenic hormones. However, it has been reported that immortalized cells in culture can be able to escape senescence and acquired ability to grow indefinitely (Kim et al. 2002). Thus, in this study, we aimed to establish a new immortalized bovine mammary epithelial cell line, which can be able to escape apoptotic and necrotic effects and to induce the same functional properties of the mammary gland cells. Both FITC Annexin V and PI were used to test the extent of apoptosis and necrosis in the hTBME cell line. Our findings showed that very few apoptotic and necrotic cells existed among the immortalized hTBME cell line. Hence, we strongly believe that our established hTBME cell line can spontaneously overcome proliferation barriers and gained immortalization. In contrast, Zhao et al. (2010a) showed in their study that many hTERT positive cell colonies could not escape from irreversible cellular senescence.
The main objective of this study is not only to establish a new model of a continues immortalized bovine mammary epithelial cell line, which can be spontaneously grown without any growth and proliferative abnormalities but also to include preservation of relatively original secretory function of natural cells of the mammary gland. To achieve this purpose in our established hTBME cell line, the ultrastructural appearance of cells was observed under TEM. Results obtained by TEM revealed that lipid droplets and secretory vesicles existed in cells, and all aggregated at the apical membrane, which matches other previous findings (Tait et al. 1990; Zhao et al. 2010b).
Besides milk fat, milk protein is another fundamental indicator of determination of the specific features of mammary gland. Expression of milk protein such as β-casein has been used as an essential sign of differentiation of mammary gland epithelial cells (Aoki 2006; Gibson et al. 1991). A combination of lactogenic hormones has been previously used to induce differentiation of cells, resulting in an increase of casein expression (Johnson et al. 2010; Riley et al. 2010). Interestingly, when we treated the hTBME cell line with insulin and prolactin, we observed that the hTBME cell line at passage 60 induced milk protein (β-casein) expression. Our established hTBME cell line showed properties as described in previous reports (Ahn et al. 1995; Huynh and Pollak 1995). These findings indicate that the hTBME cell line can be able to preserve the function of milk protein synthesis.
Another striking finding was that the established hTBME cell line exhibited a dome-like structure. Similar observations have been reported by different groups in bovine (Hu et al. 2009; Zhao et al. 2010b), porcine (Zheng and He 2010) and caprine (Tong et al. 2012). The dome formation in the established hTBME cell line occurred at a higher frequency when grown in medium containing a combination of lactogenic hormones in comparison to BME cells grown in basal growth medium. These findings offer room for speculation and then we can think about two possible explanations for that: one is that domes formation can be induced by lactogenic hormones, and the other is that domes formation are accompanied by expression within the cells of the β-casein milk protein, which is one of differentiation markers for the mammary gland during pregnancy and lactation. Taking all data presented in this study into consideration, we conclude that our established hTBME cell lines are (1) characterized morphologically and structurally, (2) maintained their growth and functional properties in culture for long periods, which mimic bovine mammary gland in vivo, and (3) appeared to be one of the most suitable models for studying function and gene expression activity of bovine mammary gland. The presented results in this study provide new methodological information that can be used in the future to study the function of the bovine mammary gland.
References
Ahn JY, Aoki N, Adachi T, Mizuno Y, Nakamura R, Matsuda T (1995) Isolation and culture of bovine mammary epithelial cells and establishment of gene transfection conditions in the cells. Biosci Biotechnol Biochem 59:59–64. doi:10.1271/bbb.59.59
Anand V, Dogra N, Singh S, Kumar SN, Jena MK, Malakar D, Dang AK, Mishra BP, Mukhopadhyay TK, Kaushik JK, Mohanty AK (2012) Establishment and characterization of a buffalo (Bubalus bubalis) mammary epithelial cell line. PLoS ONE 7:e40469. doi:10.1371/journal.pone.0040469
Aoki N (2006) Regulation and functional relevance of milk fat globules and their components in the mammary gland. Biosci Biotechnol Biochem 70:2019–2027. doi:10.1271/bbb.60142
Barber MC, Clegg RA, Finley E, Vernon RG, Flint DJ (1992) The role of growth hormone, prolactin and insulin-like growth factors in the regulation of rat mammary gland and adipose tissue metabolism during lactation. J Endocrinol 135:195–202
Bernstein P, Peltz SW, Ross J (1989) The poly(A)-poly(A)-binding protein complex is a major determinant of mRNA stability in vitro. Mol Cell Biol 9:659–670
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 16:349–352
Booth C, O’Shea JA (2002) Isolation and culture of intestinal epithelial cells. In: Freshney RI, Freshney MG (eds) Culture of epithelial cells, 2nd edn. Copyright 2002 Wiley-Liss Inc (2002) ISBNs: 0-471-40121-8 (Hardback); 0-471-22120-1 (Electronic)
Brinster RL, Allen JM, Behringer RR, Gelinas RE, Palmiter RD (1988) Introns increase transcriptional efficiency in transgenic mice. Proc Natl Acad Sci U S A 85:836–840
Capuco AV, Ellis S, Wood DL, Akers RM, Garrett W (2002) Postnatal mammary ductal growth: three-dimensional imaging of cell proliferation, effects of estrogen treatment, and expression of steroid receptors in prepubertal calves. Tissue Cell 34:143–154
Christman SA, Kong BW, Landry MM, Kim H, Foster DN (2006) Contributions of differential p53 expression in the spontaneous immortalization of a chicken embryo fibroblast cell line. BMC Cell Biol 7:27. doi:10.1186/1471-2121-7-27
Davis T, Kipling D (2005) Telomeres and telomerase biology in vertebrates: progress towards a non-human model for replicative senescence and ageing. Biogerontology 6:371–385. doi:10.1007/s10522-005-4901-4
Fu M, Chen Y, Xiong X, Lan D, Li J (2014) Establishment of mammary gland model in vitro: culture and evaluation of a yak mammary epithelial cell line. PLoS ONE 9:e113669. doi:10.1371/journal.pone.0113669
Fuchs E (1988) Keratins as biochemical markers of epithelial differentiation. Trends Genet 4:277–281
Gao K, Lu YR, Wei LL, Lu XF, Li SF, Wan L, Li YP, Cheng JQ (2008) Immortalization of mesenchymal stem cells from bone marrow of rhesus monkey by transfection with human telomerase reverse transcriptase gene. Transplant Proc 40:634–637. doi:10.1016/j.transproceed.2008.01.053
Geraghty RJ, Capes-Davis A, Davis JM, Downward J, Freshney RI, Knezevic I, Lovell-Badge R, Masters JR, Meredith J, Stacey GN, Thraves P, Vias M; Cancer Research UK (2014) Guidelines for the use of cell lines in biomedical research. Br J Cancer 111:1021–1046. doi:10.1038/bjc.2014.166
German T, Barash I (2002) Characterization of an epithelial cell line from bovine mammary gland. In Vitro Cell Dev Biol Anim 38:282–292. doi:10.1290/1071-2690(2002)038<0282:COAECL>2.0.CO;2
Gibson CA, Vega JR, Baumrucker CR, Oakley CS, Welsch CW (1991) Establishment and characterization of bovine mammary epithelial cell lines. In Vitro Cell Dev Biol 1993 Jan;29A:86.
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell Biol Int 100:57–70
Herbert BS, Wright WE, Shay JW (2002) p16(INK4a) inactivation is not required to immortalize human mammary epithelial cells. Oncogene 21:7897–7900. doi:10.1038/sj.onc.1205902
Hong HX, Zhang YM, Xu H, Su ZY, Sun P (2007) Immortalization of swine umbilical vein endothelial cells with human telomerase reverse transcriptase. Mol Cells 24:358–363
Hou M, Xu D, Bjorkholm M, Gruber A (2001) Real-time quantitative telomeric repeat amplification protocol assay for the detection of telomerase activity. Clin Chem 47:519–524
Hu H, Wang J, Bu D, Wei H, Zhou L, Li F, Loor JJ (2009) In vitro culture and characterization of a mammary epithelial cell line from Chinese Holstein dairy cow. PLoS ONE 4:e7636. doi:10.1371/journal.pone.0007636
Huynh H, Pollak M (1995) HH2A, an immortalized bovine mammary epithelial cell line, expresses the gene encoding mammary derived growth inhibitor (MDGI). In Vitro Cell Dev Biol Anim 31:25–29. doi:10.1007/BF02631334
Huynh HT, Robitaille G, Turner JD (1991) Establishment of bovine mammary epithelial cells (MAC-T): an in vitro model for bovine lactation. Exp Cell Res 197:191–199
Iannuzzi L (1994) Standard karyotype of the river buffalo (Bubalus bubalis L., 2n = 50). Report of the committee for the standardization of banded karyotypes of the river buffalo. Cytogenet Cell Genet 67:102–113
Jedrzejczak MA (2010) Bovine mammary epithelial cell culture an alternative model for study of modification and mammary gland functions. PhD dissertation, West Pomeranian University of Technology, Szczecin
Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu CP (1999) Telomerase expression in human somatic cells does not induce changes associated with a transformed phenotype. Nat Genet 21:111–114. doi:10.1038/5056
Johnson TL, Fujimoto BA, Jimenez-Flores R, Peterson DG (2010) Growth hormone alters lipid composition and increases the abundance of casein and lactalbumin mRNA in the MAC-T cell line. J Dairy Res 77:199–204. doi:10.1017/S0022029910000087
Kim H, Farris J, Christman SA, Kong BW, Foster LK, O’Grady SM, Foster DN (2002) Events in the immortalizing process of primary human mammary epithelial cells by the catalytic subunit of human telomerase. Biochem J 365:765–772. doi:10.1042/BJ20011848
Kozłowski M, Motyl T (2007) Use of three-dimensional cultures (3D) in the investigation of bovine mammary gland biology. Med Weter 63:1417–1420
Kwak S, Jung JE, Jin X, Kim SM, Kim TK, Lee JS, Lee SY, Pian X, You S, Kim H, Choi YJ (2006) Establishment of immortal swine kidney epithelial cells. Anim Biotechnol 17:51–58
Lee KM, Choi KH, Ouellette MM (2004) Use of exogenous hTERT to immortalize primary human cells. Cytotechnology 45:33–38. doi:10.1007/10.1007/s10616-004-5123-3
Moore CM, Janish C, Eddy CA, Hubbard GB, Leland MM, Rogers J (1999) Cytogenetic and fertility studies of a rheboon, rhesus macaque (Macaca mulatta) x baboon (Papio hamadryas) cross: further support for a single karyotype nomenclature. Am J Phys Anthropol 110:119–127. doi:10.1002/(SICI)1096-8644(199910)110:2<119:AID-AJPA1>3.0.CO;2-S
Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW (1999) Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nat Genet 21:115–118. doi:10.1038/5063
Morales CP, Gandia KG, Ramirez RD, Wright WE, Shay JW, Spechler SJ (2003) Characterisation of telomerase immortalised normal human oesophageal squamous cells. Gut 52:327–333
Neville MC, McFadden TB, Forsyth I (2002) Hormonal regulation of mammary differentiation and milk secretion. J Mammary Gland Biol Neoplasia 7:49–66
Ouellette MM, McDaniel LD, Wright WE, Shay JW, Schultz RA (2000) The establishment of telomerase-immortalized cell lines representing human chromosome instability syndromes. Hum Mol Genet 9:403–411
Povey RC, Osborne AD (1969) Mammary gland neoplasia in the cow. A review of the literature and report of a fibrosarcoma. Pathol Vet 6:502–512
Ramirez RD, Herbert BS, Vaughan MB, Zou Y, Gandia K, Morales CP, Wright WE, Shay JW (2003) Bypass of telomere-dependent replicative senescence (M1) upon overexpression of Cdk4 in normal human epithelial cells. Oncogene 22:433–444. doi:10.1038/sj.onc.1206046
Richert MM, Schwertfeger KL, Ryder JW, Anderson SM (2000) An atlas of mouse mammary gland development. J Mammary Gland Biol Neoplasia 5:227–241
Riley LG, Gardiner-Garden M, Thomson PC, Wynn PC, Williamson P, Raadsma HW, Sheehy PA (2010) The influence of extracellular matrix and prolactin on global gene expression profiles of primary bovine mammary epithelial cells in vitro. Anim Genet 41:55–63. doi:10.1111/j.1365-2052.2009.01964.x
Said A, Osterrieder N (2014) Equine herpesvirus type 1 (EHV-1) open reading frame 59 encodes an early protein that is localized to the cytosol and required for efficient virus growth. Virology 449:263–269. doi:10.1016/j.virol.2013.11.035
Said A, Damiani A, Osterrieder N (2014) Ubiquitination and degradation of the ORF34 gene product of equine herpesvirus type 1 (EHV-1) at late times of infection. Virology 460:11–22. doi:10.1016/j.virol.2014.05.009
Schmid E, Schiller DL, Grund C, Stadler J, Franke WW (1983) Tissue type-specific expression of intermediate filament proteins in a cultured epithelial cell line from bovine mammary gland. J Cell Biol 96:37–50
Senapathy P, Shapiro MB, Harris NL (1990) Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol 183:252–278
Serakinci N, Christensen R, Graakjaer J, Cairney CJ, Keith WN, Alsner J, Saretzki G, Kolvraa S (2007) Ectopically hTERT expressing adult human mesenchymal stem cells are less radiosensitive than their telomerase negative counterpart. Exp Cell Res 313:1056–1067. doi:10.1016/j.yexcr.2007.01.002
Shi H, Shi H, Luo J, Wang W, Haile AB, Xu H, Li J (2014) Establishment and characterization of a dairy goat mammary epithelial cell line with human telomerase (hT-MECs). Anim Sci J 85:735–743. doi:10.1111/asj.12206
Tait L, Soule HD, Russo J (1990) Ultrastructural and immunocytochemical characterization of an immortalized human breast epithelial cell line, MCF-10. Cancer Res 50:6087–6094
Talhouk RS, Neiswander RL, Schanbacher FL (1990) In vitro culture of cryopreserved bovine mammary cells on collagen gels: synthesis and secretion of casein and lactoferrin. Tissue Cell 22:583–599
Taylor-Papadimitriou J, Stampfer M, Bartek J, Lewis A, Boshell M, Lane EB, Leigh IM (1989) Keratin expression in human mammary epithelial cells cultured from normal and malignant tissue: relation to in vivo phenotypes and influence of medium. J Cell Sci 94:403–413
Tong HL, Li QZ, Gao XJ, Yin DY (2012) Establishment and characterization of a lactating dairy goat mammary gland epithelial cell line. In Vitro Cell Dev Biol Anim 48:149–155. doi:10.1007/s11626-012-9481-4
Toouli CD, Huschtscha LI, Neumann AA, Noble JR, Colgin LM, Hukku B, Reddel RR (2002) Comparison of human mammary epithelial cells immortalized by simian virus 40 T-Antigen or by the telomerase catalytic subunit. Oncogene 21:128–139. doi:10.1038/sj.onc.1205014
Vaziri H, Benchimol S (1998) Reconstitution of telomerase activity in normal human cells leads to elongation of telomeres and extended replicative life span. Curr Biol 8:279–282
Wang W, Said A, Wang Y, Fu Q, Xiao Y, Lv S, Shen Z (2015) Establishment and characterization of duck embryo epithelial (DEE) cell line and its use as a new approach toward DHAV-1 propagation and vaccine development. Virus Res 29:260–268
Wong SC, Ong LL, Er CP, Gao S, Yu H, So JB (2003) Cloning of rat telomerase catalytic subunit functional domains, reconstitution of telomerase activity and enzymatic profile of pig and chicken tissues. Life Sci 73:2749–2760
Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, Bronstein A, Chiu CP, Herron GS (1999) Human endothelial cell life extension by telomerase expression. J Biol Chem 274:26141–26148
Yudoh K, Matsuno H, Nakazawa F, Katayama R, Kimura T (2001) Reconstituting telomerase activity using the telomerase catalytic subunit prevents the telomere shorting and replicative senescence in human osteoblasts. J Bone Miner 16:1453–1464. doi:10.1359/jbmr.2001.16.8.1453
Zavizion B, Gorewit RC, Politis I (1995a) Subcloning the MAC-T bovine mammary epithelial cell line: morphology, growth properties, and cytogenetic analysis of clonal cells. J Dairy Sci 78:515–527. doi:10.3168/jds.S0022-0302(95)76662-0
Zavizion B, van Duffelen M, Schaeffer W, Politis I (1995b) Use of microinjection to generate an immortalized bovine mammary cell line with both epithelial and myoepithelial charcteristics. Methods Cell Sci 17:271–282
Zavizion B, van Duffelen M, Schaeffer W, Politis I (1996) Establishment and characterization of a bovine mammary epithelial cell line with unique properties. In Vitro Cell Dev Biol Anim 32:138–148
Zhao CF, Hu HY, Meng L, Li QQ, Lin AX (2010a) Immortalization of bovine mammary epithelial cells alone by human telomerase reverse transcriptase. Cell Biol Int 34:579–586. doi:10.1042/CBI20100006
Zhao K, Liu HY, Zhou MM, Liu JX (2010b) Establishment and characterization of a lactating bovine mammary epithelial cell model for the study of milk synthesis. Cell Biol Int 34:717–721. doi:10.1042/CBI20100023
Zheng YM, He XY (2010) Characteristics and EGFP expression of porcine mammary gland epithelial cells. Res Vet Sci 89:383–390. doi:10.1016/j.rvsc.2010.03.023
Acknowledgements
The work was supported by the Key Program of the National Science Foundation of China [Grant No: 2008AA101006, China], and supported by grants from the National Natural Science Foundation of China (Grant Nos. 31572492, 31072109), the Natural Science Foundation of Tianjin, China (Grant No. 12JCZDJC22100), the Veterinary Biotechnology Scientific Research Innovation Team of Tianjin, China (Grant No. TD12-5019), and the Veterinary “Leading Talent Culture Project” of Tianjian, China.
Authors’ contribution
JL and AS contributed equally to this work by designing, carrying out experiments, analyzing and interpreting the data. XG and WW contributed to analyze and interpret the data of this work. AS, JL, YZ and TJ contributed to the drafting of the manuscript, revising it critically, and giving final approval of the version to be published. All authors read and approved the final manuscript. All contributors who do not meet the criteria for authorship should be listed in an acknowledgements section.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical standard
Bovine mammary gland tissue was obtained from a local slaughterhouse in Shannxi, Xian, China. Animals were not killed for this scientific research, therefore, no ethical approval was needed for tissue collection.
Additional information
Ji-Xia Li and Abdelrahman Said have contributed equally to this work.
Rights and permissions
About this article

Cite this article
Li, JX., Said, A., Ge, XG. et al. Development and validation of immortalized bovine mammary epithelial cell line as an in vitro model for the study of mammary gland functions. Cytotechnology 70, 67–82 (2018). https://doi.org/10.1007/s10616-017-0114-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-017-0114-3