
Abstract
In this study, highly purified hair follicle stem cells from Arbas Cashmere goat (gHFSCs) were isolated using enzyme digestion and adhesion to type IV collagen. The biological characteristics of the gHFSCs were identified by morphological observation, growth curve, markers assay and differentiation in vitro. The gHFSCs were in small cell size with typical cobblestone morphology, good adhesion and high refractive index. Immunocytochemistry staining showed the cells were expressing Krt15, Krt19, CD34, Itgβ1 and Krt14. Cell growth curve indicated that cultured gHFSCs had strong proliferation ability. Krt14 and CD34 were high expressed at the mRNA level, respectively, 39.68 and 24.37 times of the Cashmere goat keratinocytes, and krt15 expression was 5.62 times and itgβ1 expression was 1.81 times higher (p < 0.01). Western blot detected the expression of all the above markers. After osteogenic induction, the cells were positive for Von Kossa staining and expressed Osteocalcin. Sulfated proteoglycans in cartilaginous matrices were positively stained by Alcian blue after chondrogenic induction and COL2A1 was expressed. In myogenic induction, Hoechst 33342 staining evidenced cytoplasm fusion and positive expression of MyoG was detected by immunocytochemistry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hair follicles (HF) not only have important biological functions such as prevention of skin tissue damage, important sensory and immunologic functions, but also contain diverse stem cells for infinite proliferation, differentiation, hair growth regulation, and skin homeostasis maintenance. Therefore some scientists described HF as a stem cell zoo (Jaks et al. 2010; Schneider et al. 2009). Hair follicle stem cells (HFSCs) are required to initiate, maintain and renew the continuously HF cycle. It is well-established that HFSCs are located predominantly at the HF bulge. Ever since their identification in mice (Cotsarelis et al. 1990) the biology of HFSCs has become a promising frontier in the dermatology field and regenerative medicine. Previous studies have indicated that HFSCs have a wide range of potential to differentiate into neurogenic, adipogenic, myogenic, osteogenic and chondrogenic lineages after appropriate induction in vitro (Amoh et al. 2005; Bajpai et al. 2012; Liu et al. 2010).
The Inner Mongolia Arbas Cashmere goat (Capra hircus) is a local cashmere and meat dual-purpose breed, which produces fine, soft, quality cashmere known as “soft-gold” or “fiber gems”. The average down yield is about 740–850 g, with an average down diameter between 14.3 and 15.6 µm. The cashmere length is between 66 and 76 mm (Liu et al. 2006). The Arbas Cashmere goats have two different types of hair follicles, primary and secondary ones (Zhu et al. 2014) with obvious hair cycle, and is promptly becoming a popular model for HF morphogenesis research.
However, domestic animal HFSCs are less studied compared to human and murine counterparts and there are few studies about Arbas Cashmere goat HFSCs (gHFSCs). In this study, we isolated gHFSCs and detected most commonly used HFSCs markers, such as Keratin15 (Krt15), Keratin19 (Krt19), CD34, and integrinβ1 (Itgβ1), outer root sheath special marker keratin14 (Krt14) by immunocytochemistry staining, quantitative real-time polymerase chain reaction (Q-PCR) and Western blot analysis. We evaluated the proliferation capacity of gHFSCs at different passages by cell growth curve. Osteogenic, chondrogenic and myogenic induction was performed to test the differentiation potential of the gHFSCs. We believe that gHFSCs research has vital significance on improving cashmere quality and production.
Materials and methods
Animals: Adult Arbas Cashmere goat was obtained from the Inner Mongolia YIWEI white Cashmere Goat Farm. All studies were performed with the approval of the Experimental Animal Committee of the Inner Mongolia University.
Reagents: Dulbecco’s modified Eagle’s medium (DMEM/F-12) (SH30023, Hyclone, Logan, UT, USA), FBS (TBD31 HB, tbd, Tianjin, China), collagen IV (C5533, Sigma), EGF (E1257, Sigma), insulin (I0516, Sigma), hydrocortisone (H0888, Sigma), penicillin–streptomycin mixture (SV30010, Hyclone), Phosphate Buffered Saline (PBS) (SH30028, Hyclone), Dispase (D4818, Sigma), TrypLE™ Express Enzyme (1621388, Gibco, Grand Island, NY, USA), dimethyl sulfoxide (DMSO) (042-21765, Wako Pure Chemicals, Osaka, Japan), paraformaldehyde (P6148, Sigma), TritonX-100 (9002-93-1, Sigma), 40,6-diamidino-2-phenylindole (DAPI) (D8417, Sigma), RIPA lysis buffer (2089131, Millipore, Billerica, MA, USA), β-glycerophosphate disodium salt hydrate (G5422, Sigma), Hoechst 33342 (14533, Sigma), L-ascorbic acid (A7506, Sigma), dexamethasone (D4902, Sigma), TGFβ-1 (T7039, Sigma), horse serum (16050-122, Gibco). Primary antibodies against integrinβ1 (ab155145), keratin14 (ab181595), keratin15 (ab111448), keratin19 (ab15463), CD34 (ab81289), MyoG (ab77232), and secondary antibodies goat anti rabbit IgG AlexaFlour®488 (ab150077) and horseradish peroxidase-linked goat anti rabbit IgG (ab6721) were all purchased from Abcam (Cambridge, MA, USA). RNAiso Plus (9109), Prime Script™RT reagent kit (RR047A), and SYBR®Premix Ex Taq (RR820A) were all from Takara (Dalian, China).
Coating the dishes
Collagen IV was diluted according to the manufacturer’s instruction then coated the six-well dish (1 mL/well). The plates were placed in the clean bench for 30 min, and then put into the incubator for more than one hour. The coating solution was retrieved, and the plates were washed three times with PBS before cell culture.
HF adhesion and culture of the primary gHFSCs
Clip 1 cm × 1 cm skin from neck of the adult Arbas Cashmere goat and disinfected in 75 % ethanol. The skin was cut into 5 mm × 5 mm blocks using ophthalmic scissors, and washed with PBS three times. The samples were treated with 0.25 % dispase II at 4 °C overnight. HFs were separated from the connective tissue using a syringe needle under stereomicroscope, and cultured in medium DMEM/F12 supplemented with 2 % FBS, 1 % penicillin–streptomycin mixture, 10 ng/mL EGF, 10 ng/mL insulin and 0.4 µg/mL hydrocortisone at 37 °C with 5 % CO2. After tissue adherence, medium was changed every 3 days and cell growth was observed microscopically.
Purification of the gHFSCs
The gHFSCs were purified using fast adhesion to collagen IV using a modified previously described method (Bickenbach and Chism 1998). Primary cells were digested and centrifuged at 1500 rpm for 5 min, then pipetted into single cell suspension and cultured in collagen IV treated six-well dishes for 20 min. The non-adherent cells were discarded with the medium and the adherent cells were further cultured. Medium was changed every 3 days and the purity of the cells was detected by CD34 FACS (fluorescence activated cell sorting). After being harvested, gHFSCs were fixed in 4 % paraformaldehyde for 40 min and permeabilized with 0.1 % Triton X-100 in 10 % goat serum for 10 min on ice then incubated sequentially with unconjugated primary antibodies for 1 h and FITC-conjugated secondary antibody for 40 min on ice. FACS was performed by BD FACS Canto (Diva7.0) (San Jose, CA, USA) and analyzed by FlowJo.
Passage, cryopreservation and cell growth curve
We seeded 2 × 105 cells in each 6-well dish and passaged them when they reached 90 % of confluence. The gHFSCs were digested with TrypLE™Express Enzyme, centrifuged, and then suspended. Part of the cells was passaged and cultured at 37 °C with 5 % CO2 and medium was changed every 3 days. The rest of the cells was added into the mixture of 90 % FBS-10 % DMSO as a cryoprotectant, and placed at −80 °C overnight then put into liquid nitrogen tank for long-term storage. The gHFSCs of 3rd passage (P3), 6th passage (P6) and 10th passage (P10) in good condition were seeded in 24-well plates at a density of 1 × 104 cells/mL and cell counts were performed every 24 h by hemocytometer. Three wells were counted for each time point, and the mean cell number was calculated to plot the cell growth curve.
Immunocytochemistry
P5 gHFSCs were cultured on 24-well dishes with cover slips at a density of 1 × 105 cells/well. When the cells reached 70 % of confluence, the medium was discarded, then the cells were washed with PBS and fixed with 4 % paraformaldehyde at room temperature for 15 min. Cells were permeabilized with 0.1 % Triton X-100 in PBS for 10 min. After washing (3×), the specimens were blocked with 10 % goat serum for 1 h at room temperature. Primary antibodies against Itgβ1 (1:300), Krt14 (1:300), Krt15 (1:300), Krt19 (1:300) and CD34 (1:300) were added separately to the samples and incubated at 37 °C for 1 h. Negative control was incubated with 10 % goat serum instead of primary antibodies. After washing (3×), cells were incubated with secondary antibody (1:500) in the dark for 45 min. Finally, the nuclei were counterstained with DAPI (1:1000) for 5 min in the dark. At least three replicates were performed for each sample. The cells were visualized using a confocal microscope (A1, Nikon, Tokyo, Japan).
Real-time quantitative PCR
Itgβ1, CD34, krt14, krt15 mRNA expression levels in gHFSCs were measured using Q-PCR. All primer sequences were determined using established GenBank sequences, which are listed in Table 1 (no sequence of Capra hircus krt19 was found in the GenBank). The gHFSCs were lysed by RNAiso Plus reagent to extract total RNA then reverse transcribed into cDNA using the PrimeScript™RT reagent kit according to the manufacturer’s instructions. The Q-PCR reaction system comprised 2 × SYBR Green Mix (10 μL), primer mix (1 μL), template (1 μL), and ddH2O (8 μL). Q-PCR parameters were as follows: 95 °C for 30 s followed by 40 cycles of 95 °C for 30 s and 60 °C for 30 s. The GAPDH gene was used as a reference gene. The relative mRNA expression level of each gene from triplicate experiments was calculated using the 2−ΔΔCt method (Livak and Schmittgen 2001).
Western blot
Total cellular extracts of gHFSCs were obtained for the Western blot using RIPA lysis buffer, and protein concentrations of the cell lysates were determined by the spectrophotometer method. Aliquots of cell lysates containing 12 μg proteins were electrophoretically separated by 12 % SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. After incubating for 2 h with blocking buffer (TBST containing 5 % skimmed milk), the membranes were probed with primary antibodies against Itgβ1, CD34, Krt14, Krt15, Krt19 overnight at 4 °C. After washing (3×) with TBST, the membranes were incubated with horseradish peroxidase-linked goat anti-rabbit IgG at room temperature for 1 h, then protein bands were detected using enhanced chemiluminescence.
Osteogenic, chondrogenic and myogenic induction
For osteogenic differentiation, P5 gHFSCs were cultured for 3 weeks in induction medium DMEM/F12 supplemented with 10 % FBS, 0.2 mM ascorbic acid, 100 nM dexamethasone and 10 mM β-glycerophosphate. Osteogenic differentiation was confirmed by positive Von Kossa staining of mineralized matrix and the expression of Osteocalcin. For chondrogenic differentiation, cells were cultured for 3 weeks in DMEM/F12 supplemented with 10 % FBS, 100 nM dexamethasone, 0.2 mM ascorbic acid and 100 nM human TGFβ-1. Chondrogenic differentiation was confirmed by positive Alcian blue staining of cartilaginous matrices and the expression of COL2A1. For myogenic differentiation, cells were cultured in DMEM/F12 supplemented with 2 % horse serum. When the cells appeared with a tube-like morphology, they were stained with 5 mg/L Hoechst 33342 to observe cytoplasm fusion. After 3 weeks, cells were stained using antibody against MyoG to confirm myogenic differentiation. Control group cells were cultured in DMEM/F12 supplemented with 10 % FBS for 3 weeks in parallel. The primer sequences of Osteocalcin and COL2A1 are listed in Table 2.
In this study, each experiment was repeated at least 3 times. All data are presented as mean and standard error, and analyzed using the SPSS 17.0 for windows. A value of p less than 0.05 (p < 0.05) was considered statistically significant.
Results
Morphologic characteristics and identification of gHFSCs
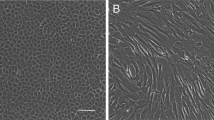
On day 3 of HF culture, a few cells had migrated from the hair follicle bulge (Fig. 1a). On day 5, cell number was increased and most of the cells displayed cobblestone morphology and were closely aligned, which is typical of epithelial cells (Fig. 1b). The gHFSCs (primary passage and then second passage) were purified by fast adhesion to collagen IV. After purification, approximately 90 % of the cells showed typical morphology of hair follicle stem cells, including a cobblestone and nest appearance, stereoscopic impression, high refractive index, high colony-forming ability, good adhesion ability, small cell size, centralized, round and large nuclei with more than two nucleoli (Fig. 1c) and CD34 FACS positive rate was 99.9 % (Fig. 1d). Immunocytochemistry staining showed positive expression of Krt15, Krt19, CD34, Itgβ1 and Krt14 in the gHFSCs (Fig. 2).

Morphology of gHFSCs. a Adherent culture of hair follicle, b primary gHFSCs from HF culture, c after purification, d FACS analysis of CD34: the upper panel presents cells only incubated with the secondary antibody (control), the lower panel presents cells incubated with anti-CD34 antibody. Vertical axis: Cell number, horizontal axis: Channel number, e passage 15 gHFSCs. Scale bars 100 μm

Immunocytochemistry staining. Scale bars 100 µm
Proliferation of the gHFSCs of different passages
The gHFSCs of P3, P6 and P10 were in the latent phase on days 1–3, proliferating slowly. Stem cell colonies appeared on day 3, and from day 4 the cells started proliferating rapidly and entered logarithmic phase. Proliferation then slowed down gradually into the plateau phase on day 8 (Fig. 3).

Growth curve of different passage gHFSCs
Relative mRNA expression level of HFSCs markers
At the mRNA level, the expression of krt14, CD34, krt15 and itgβ1 in gHFSCs were, respectively, 39.68, 24.37, 5.62 and 1.81 times of their expression in Cashmere goat keratinocytes, that is krt14 and CD34 were highly expressed, with medium expression for krt15, and the expression of itgβ1 was low (p < 0.01).
Protein expression of the HFSCs markers
Western blot also detected the protein expression of Krt15, Krt19, CD34, Itgβ1 and Krt14 in the gHFSCs with α-Tubulin as an internal reference protein (Fig. 4b).

The mRNA and protein expression of gHFSCs markers. a Relative mRNA expression level of gHFSCs markers, b protein expression of gHFSCs markers
Confirmation of osteogenic, chondrogenic and myogenic differentiation
After Von Kossa staining, we found the cells were stained black through microscopic observation in the osteogenic induction group, and were not stained in control group; this means that mineralized nodules were formed in osteogenic induced gHFSCs (Fig. 5a, b). The expression of Osteocalcin was detected by gel electrophoresis (Fig. 5c). After chondrogenic differentiation, the cells were stained light blue after Alcian blue staining but control group was not stained; this showed that cartilaginous matrices and the distinct lacuna structure of cartilage were formed in the chondrogenic induced gHFSCs (Fig. 6a, b). COL2A1 expression was detected (Fig. 6c). Moreover, gHFSCs began to fuse after myogenic induction for 5 days, leading to obvious myotube cell formation. Hoechst 33342 staining revealed the presence of multiple nuclei in the same myotube (Fig. 7a), and immunohistochemistry staining demonstrated that the cells were MyoG positive and control cells were MyoG negative (Fig. 7b–d). These results indicated that gHFSCs can be differentiated into osteoblasts, chondroblasts, and myoblasts lineage under appropriate culture condition.

Identification of osteogenic differentiation. a Induction group, b control group, c expression of Osteocalcin (294 bp) mRNA, M 100 bp marker, 1 induction group, 2 control group, 3 normal cultured gHFSCs. Scale bars 100 µm

Identification of chondrogenic differentiation. a Induction group, b control group, c expression of COL2A1 (433 bp) mRNA, M 100 bp marker, 1 induction group, 2 control group, 3 normal cultured gHFSCs. Scale bars 100 µm

Identification of myogenic differentiation. a Hoechst 33342 staining, b, c induction group stained for MyoG expression (immunofluorescence staining), d control group stained for MyoG (immunofluorescence staining). Scale bars 100 μm
Discussion
Microdissection, enzymatic digestion, and FACS were most routinely used techniques for isolating potential stem cells from the HF. Microdissection is commonly used in isolating dermal papilla cells. It preserves the whole tissues in situ, thereby increasing the efficiency of cell isolation. However, it is quite laborious and requires experienced technicians. Enzymatic digestion can isolate HF from the surrounding dermis and stem cells from the HF, and it is impossible to determine the cell types that are obtained. FACS is a highly effective technique that can perform identification, classification and quantification for one to four specific cells at the same time. It is applied to screening HFSCs using specific antibodies (Mistriotis and Andreadis 2013). So far, lack of reliable stem cell markers restrains use of this method in sorting HF stem cells. Microdissection, the current method for their isolation and culture, alone cannot isolate HFSCs with high purity. In addition, epidermal stem cells show good adhesion ability compared to other cell types, with a faster adhesion (within 10–20 min) to collagen IV, laminin, and extracellular matrix (Jones and Watt 1993). In this work, we combined enzymatic digestion with microdissection to isolate gHFSCs, and differential adhesion to collagen IV to purify gHFSCs, voided disadvantages of methods mentioned above. This adhesion method was used in isolating rat HFSCs (Quan et al. 2014), and we consider it could also be applied to isolate murine and human HFSCs.
Most widely used markers in identifying murine and human HFSCs are Krt15, Krt19, CD200, CD34, PHLDA, and Itgβ1 and Itgα6 are considered as potential specific HFSCs markers (Ernst et al. 2013; Kloepper et al. 2008; Purba et al. 2014). However, the biological function of these genes is still obscure. In the murine bulge, cells expressing Krt15 are with high clonality and can regenerate all epithelial lineages (Bose et al. 2013). Krt19 is another crucial HFSC marker which is expressed in the bulge and suprabulbar ORS (Commo et al. 2000). In the murine bulge, CD34 is co-localized with Krt15, and CD200 is expressed aside Krt15 in the human bulge, therefore, they could be effective markers to identify supposed HFSCs (Poblet et al. 2006). PHLDA1 is an evolutionarily conserved proline-glutamine rich nuclear protein. DNA microarray analysis in human HF found expression of PHLDA1 and its receptor in the bulge region. PHLDA1 is also used to discriminate other cells so it is not featured to HFSCs (Ohyama et al. 2006). Krt14 is an outer root sheath special marker (Vidal et al. 2005).
In this study, Krt15, Krt19, CD34, Krt14 and itgβ1 were used as markers for gHFSCs, and immunofluorescence staining showed that the gHFSCs were positive for all five markers. The Q-PCR result showed that in gHFSCs, the expression of krt14 and CD34 was significantly high, with medium expression of krt15 and lower expression of Itgβ1 (p < 0.01). We isolated the gHFSCs from telogen HF. Itgβ1 was described to be associated with epithelial progenitor cell proliferation (Ernst et al. 2013). We isolated gHFSCs from telogen gHF, therefore, low expression of Itgβ1 was partially due to the quiescent state of the cells. Western blot demonstrated the differential expression of gHFSCs markers at protein level as well. These results indicated that our isolated cells are consistent with the HFSCs characteristics. The small cell size and good adhesion ability of gHFSCs made digestion longer and more difficult compared with other types of cells. It has been reported that the HFSCs can be cultured in medium supplemented with 10 % FBS (Guo et al. 2011; Quan et al. 2014). In our study, most of the cells turned into spindle-like morphology when cultured with medium containing 10 % FBS. Interestingly, when cultured in medium only containing 10 % FBS, the cells grew into spheres (Sup.1). Epithelial cells are exquisitely sensitive to calcium, and it is essential to carefully control the calcium levels of the medium (Nowak and Fuchs 2009). Therefore, we suspected that the morphological changes of the cells was related to the use of unchelated FBS and the differentiation potential of gHFSCs. Previous studies have indicated that the hair follicle is a readily accessible mini-organ within the skin that contains stem cells with notably broad differentiation potential. In our study, positive results for osteogenic, chondrogenic and myogenic differentiation further confirmed the in vitro differentiation ability of gHFSCs. In comparison with other adult stem cells, HFSCs are easier to obtain with minimal trauma. Therefore, HFSCs could become a novel seed cell with more advantages for tissue engineering and clinical medicine.
Conclusions
High purity gHFSCs can be obtained using enzymatic digestion with microdissection and fast adhesion to collagen IV. The cultured cells had good proliferative capacity and were positive for HFSCs markers. Q-PCR and Western blot showed mRNA and protein expression of HFSCs markers. gHFSCs can differentiate into osteogenic, chondrogenic and myogenic lineages under appropriate culture condition. HFSCs could be a preferred, novel cell source for tissue engineering and regenerative medicine.
References
Amoh Y, Li L, Katsuoka K, Penman S, Hoffman RM (2005) Multipotent nestin-positive, keratin-negative hair-follicle bulge stem cells can form neurons. Proc Natl Acad Sci USA 102:5530–5534
Bajpai VK, Mistriotis P, Andreadis ST (2012) Clonal multipotency and effect of long-term in vitro expansion on differentiation potential of human hair follicle derived mesenchymal stem cells. Stem Cell Res 8:74–84
Bickenbach JR, Chism E (1998) Selection and extended growth of murine epidermal stem cells in culture. Exp Cell Res 244:184–195
Bose A, Teh M-T, Mackenzie IC, Waseem A (2013) Keratin k15 as a biomarker of epidermal stem cells. Int J Mol Sci 14:19385–19398
Commo S, Gaillard O, Bernard BA (2000) The human hair follicle contains two distinct K19 positive compartments in the outer root sheath: a unifying hypothesis for stem cell reservoir? Differentiation 66:157–164
Cotsarelis G, Sun T-T, Lavker RM (1990) Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 61:1329–1337
Ernst N, Yay A, Bíró T, Tiede S, Humphries M, Paus R, Kloepper JE (2013) β1 Integrin signaling maintains human epithelial progenitor cell survival in situ and controls proliferation, apoptosis and migration of their progeny. PLoS One 8:e84356
Guo Z, Draheim K, Lyle S (2011) Isolation and culture of adult epithelial stem cells from human skin. JoVE 49. doi:10.3791/2561
Jaks V, Kasper M, Toftgård R (2010) The hair follicle—a stem cell zoo. Exp Cell Res 316:1422–1428
Jones PH, Watt FM (1993) Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell 73:713–724
Kloepper JE, Tiede S, Brinckmann J, Reinhardt DP, Meyer W, Faessler R, Paus R (2008) Immunophenotyping of the human bulge region: the quest to define useful in situ markers for human epithelial hair follicle stem cells and their niche. Exp Dermatol 17:592–609
Liu X, Zhang P, Liu S (2006) Breed conservation and utilization of Inner Mongolia Arabs Cashmere goat China. Anim Husb Vet Med 32:34–36
Liu JY, Peng HF, Gopinath S, Tian J, Andreadis ST (2010) Derivation of functional smooth muscle cells from multipotent human hair follicle mesenchymal stem cells. Tissue Eng Part A 16:2553–2564
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 25:402–408
Mistriotis P, Andreadis ST (2013) Hair follicle: a novel source of multipotent stem cells for tissue engineering and regenerative medicine. Tissue Eng Part B Rev 19:265–278
Nowak JA, Fuchs E (2009) Isolation and culture of epithelial stem cells. Methods Mol Biol 482:215–232. doi:10.1007/978-1-59745-060-7_14
Ohyama M, Terunuma A, Tock CL, Radonovich MF, Pise-Masison CA, Hopping SB, Brady JN, Udey MC, Vogel JC (2006) Characterization and isolation of stem cell-enriched human hair follicle bulge cells. J Clin Investig 116:249
Poblet E, Jimenez F, Godinez J, Pascual-Martín A, Izeta A (2006) The immunohistochemical expression of CD34 in human hair follicles: a comparative study with the bulge marker CK15. Clin Exp Dermatol 31:807–812
Purba TS, Haslam IS, Poblet E, Jiménez F, Gandarillas A, Izeta A, Paus R (2014) Human epithelial hair follicle stem cells and their progeny: current state of knowledge, the widening gap in translational research and future challenges. BioEssays 36:513–525
Quan R, Zheng X, Ni Y, Xie S, Li C (2014) Culture and characterization of rat hair follicle stem cells. Cytotechnology 1–8. doi:10.1007/s10616-014-9807-z
Schneider MR, Schmidt-Ullrich R, Paus R (2009) The hair follicle as a dynamic miniorgan. Curr Biol 19:R132–R142
Vidal VP, Chaboissier MC, Lützkendorf S, Cotsarelis G, Mill P, Hui CC, Ortonne N, Ortonne JP, Schedl A (2005) Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr Biol 15:1340–1351
Zhu B, Xu T, Zhang Z, Ta N, Gao X, Hui L, Guo X, Liu D (2014) Transcriptome sequencing reveals differences between anagen and telogen secondary hair follicle-derived dermal papilla cells of the Cashmere goat (Capra hircus). Physiol Genomics 46:104–111
Acknowledgments
We are grateful to the Inner Mongolia YIWEI white Cashmere goat farm that provided us the goats for this study. This project was supported by the Generation of Induced Pluripotent Stem Cells from Cashmere Goat [Grant No. 2011JQ03].
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, N., Dong, Z., Tao, L. et al. Isolation and characterization of hair follicle stem cells from Arbas Cashmere goat. Cytotechnology 68, 2579–2588 (2016). https://doi.org/10.1007/s10616-016-9981-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-016-9981-2