
Abstract
Cabrio Plus, a commercial fungicide, is used in agriculture as the control agent for a broad spectrum of diseases including black dot, early blight, late blight and powdery mildew. This study aimed to evaluate the genotoxicity of commercial formulation of Cabrio Plus which has been inadequately evaluated. The genotoxic potential of Cabrio Plus in isolated human peripheral blood lymphocytes was measured by means of an alkaline version of the comet assay (pH > 13) and in whole blood by use of the in vitro micronucleus test. Cabrio Plus induced a statistically significant increase in DNA damage assessed with the in vitro micronucleus assay and the comet assay. Cabrio Plus also induced cytotoxicity in a dose-dependent manner with the in vitro micronucleus assay. It can be concluded that a commercially available pesticide formulation, Cabrio Plus, has the ability to cause DNA damage and cytotoxicity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The demand for increased crop yields and reduced post-harvest loss encourages agricultural producers to make extensive use of pesticides (Moretti et al. 2002). For similar reasons, the use of pesticides in combating pests is indispensable in comparison with other methods. Pesticides are toxic chemicals designed to kill and/or repel pests (U.S. EPA 2005). Many pesticides, as marketed and used, consist of two major components (U.S. EPA 2006), i.e. the active ingredients that prevent, destroy, and repel pests (U.S. EPA 1997), and the inert ingredients, which may have biological activity of their own and thus the potential to cause toxic effects on human health (Cox and Surgan 2006).
Numerous studies have shown the toxic potential of pesticide formulations, which may mean that inert ingredients enhance the toxicity of the active ingredient(s). However, an inert ingredient may also increase persistence and off-target the pesticides. Approximately 3000 inert ingredients with different toxic potential, half of them posing a moderate risk, have been used for many years (Cox and Surgan 2006). Before marketing and use of a pesticide, its formulation needs to be fully assessed. In order to register a pesticide in the United States, 20 toxicological tests are required, while only seven short-term acute toxicity tests are used for pesticide formulation. Other tests are performed only on the active ingredients (Cox and Surgan 2006). Furthermore, significant aspects of pesticide toxicity, including cancer and genetic damage, are conducted only on the active ingredients (Cox and Surgan 2006). Specifically, the genetic damage of active ingredients is evaluated through specific tests such as chromosomal aberration assay and the bacterial reverse gene mutation test. Growing scientific evidence has shown the inadequacy of testing pesticide formulations. In this study, we used the in vitro micronucleus test (IVMNT) and the alkaline version of the comet assay (pH > 13) to provide indicators of genotoxicity, in order to evaluate the commercial formulation of Cabrio Plus (CP).
The comet assay is a simple, rapid and sensitive method for measuring DNA breaks in a small number of cells (Dhawan and Anderson 2009) and is not a routine test for the registration of agrochemicals. In addition to providing data on the effect of genotoxic exposure in human populations, the comet assay has yielded a great deal of fundamental information on the mechanisms of genotoxicity and cellular responses to DNA damage. The alkaline version of the comet assay, detecting low levels of DNA damage, is capable of assessing DNA single-strand breaks (SSB), alkali-labile sites (ALS), DNA–DNA/DNA–protein cross-linking and SSB associated with incomplete excision repair sites (Tice et al. 2000). Micronuclei (MN) originate from chromosome fragments or whole chromosomes that lag behind at the anaphase during nuclear division (Fenech 2007). The micronucleus assay has evolved as a comprehensive method to measure chromosome breakage, DNA mis-repair, chromosome loss, non-disjunction, necrosis, apoptosis and cytostasis (Fenech 2007).
The aim of present study was to evaluate the genotoxic potential of a commercial formulation of pesticide, Cabrio Plus, on human peripheral blood lymphocytes. We employed the comet assay and in vitro micronucleus test (IVMNT) to measure the genotoxicity and used the cytokinesis-block proliferation index (CBPI) to measure cytotoxicity.
Materials and methods
Chemicals
Cabrio Plus (CP) is a broad-spectrum fungicide composed of 55 % metiram and 5 % pyraclostrobin as the active ingredients and is sold as a commercial formulation. CP was purchased from a pesticide dealer in Canakkale, Turkey. Chemicals and reagents used in the comet assay were purchased from the following suppliers: low-melting agarose (LMA), normal-melting agarose (NMA), ultrapure ethylenediamine tetraacetic acid disodium salt dehydrate (EDTA) from Invitrogen (Carlsbad, CA, USA), sodium chloride (NaCl), RPMI 1640 culture medium, mitomycin C (MMC), and cytochalasin-B from Sigma (Seelze, Germany); ultrapure Tris and phosphate-buffered saline (PBS) from Sigma; sodium hydroxide (NaOH), N-Lauroylsarcosin, dimethyl sulfoxide (DMSO), hydrogen peroxide (H2O2), potassium chloride (KCl), and Triton-X from Merck (Darmstadt, Germany); ethidium bromide from AppliChem (Darmstadt, Germany); Ficoll–Paque PREMIUM from GE Healthcare (Uppsala, Sweden), and phytohaemagglutinin from Biological Industry (Kibbutz Beit-Haemek, Israel).
Blood samples and lymphocytes isolation for comet assay
For each experiment, heparinized whole blood was collected by venipuncture from one male and one female donor, with their consent. Both donors were non-smokers, they were not exposed to radiation or pesticides, and had not used medicine for medical care. The comet assay was performed according to a method previously described, with minor modifications (Dhawan et al. 2003). Lymphocytes were isolated on a Ficoll–Paque density gradient (Bøyum 1976) and washed with culture medium containing 80 % RPMI 1640 and 20 % fetal calf serum. The lymphocytes were suspended in a total volume of 1 ml and each culture flask contained ~106 cells. A 0.01–5-µg/ml dose range of CP was investigated. The cells were incubated for 1 h at 37 °C in a shaking incubator. After the incubation period, treated cells were washed three times with cold PBS to remove fungicide residue. Two parallel cultures (duplicate cultures) for each individual were set up for selected concentrations of the fungicides. A concurrent negative and positive control was included for each experiment. CP was dissolved in RPMI 1640 medium, which was added to the negative-control cultures. Each experiment included a culture of cells treated with 50 µM of H2O2 an ice-cold flat tray for 5 min, as a positive control.
Slide preparation
In the present study, the alkaline version of the comet assay was carried out (Singh et al. 1988). Frosted microscope slides were covered with 1.25 % NMA in double-distilled water. The covered slides were kept at room temperature for 1 day, thus allowing the agarose to solidify. A total of 10,000–20,000 cells were mixed with 70 µl of 1 % LMA at 37 °C and spread out on the pre-coated slide with a cover slip. The mixture of cells and LMA was maintained on an ice-cold flat tray during five min for the agarose to solidify. After removing the cover slip, slides were immersed in cold lysing solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, 1 % N-lauroylsarcosin, pH 10) with 10 % DMSO. Triton-X (1 %) was added 1 h before use at 37 °C in the dark.
Electrophoresis
Slides were removed from the lysing solution and placed in a horizontal electrophoresis tank. The tank was filled with electrophoresis solution (4 °C) composed of 1 mM Na2EDTA and 300 mM NaOH (pH > 13). The level of electrophoresis solution was adjusted depending on preferred V and mA. The slides were left in the cold electrophoresis solution for 20 min for unwinding of DNA and expression of alkali-labile damage. Electrophoresis was performed at 4 °C for 20 min using 24 V (~0.7 V/cm) and 300 mA (Power supply, Owl, OSP300-2Q, Thermo Fisher, Waltham, MA, USA). After the electrophoresis, the slides were removed and put into double-distilled water in a coupling jar for 2–3 min. Tris buffer (0.4 M Tris, pH 7.5) was then added onto the slides to neutralize the alkali condition. The slides were left in the neutralizing buffer for 5 min. This step was repeated three times. After neutralization, the slides were kept in cold 100 % ethanol for fixation during 20 min. The slides were dried at room temperature and stored in a dry box.
Staining and slide scoring
The dried slides were rehydrated with cold distilled water for 30 min, stained with 65 µl of 20 µM ethidium bromide (EtBr) for 4–5 min, and washed in cold distilled water. The slides were examined at 400× magnification with a fluorescence microscope (Axiostar Plus®, Carl Zeiss, Gottingen, Germany). On each slide, 200 randomly selected cells were analyzed for each treatment to evaluate DNA damage. DNA damage was assessed by visual scoring of the comets, as proposed by Collins (2004). The comets were assigned to one of five classes from 0 to 4: 0 (no visible tail) to 4 (almost all DNA in tail), depending on the intensity of the comet tail. The arbitrary unit (AU) was calculated for each concentration by use of the following equation: \( (\% \,{\text{of}}\,{\text{cells}}\,{\text{in}}\,{\text{class}}\,0){\text{x}}0 + (\% \,{\text{of}}\;{\text{cells}}\,{\text{in}}\,{\text{class}}\,1){\text{x}}1 + (\% \,{\text{of}}\,{\text{cells}}\,{\text{in}}\,{\text{class}}\,2){\text{x}}2 + (\% \,{\text{of}}\;{\text{cells}}\,{\text{in}}\,{\text{class}}\,3){\text{x}}3 + (\% \,{\text{of}}\,{\text{cells}}\,{\text{in}}\,{\text{class}}\,4){\text{x}}4. \)
Whole blood lymphocyte cultures and pesticide treatment for the in vitro micronucleus test
The IVMNT was performed according to Fenech’s method with minor modifications (Fenech 2007). Each culture contained 0.5 ml whole blood, 4 ml RPMI 1640 (Sigma), 1 ml fetal calf serum, and 0.2 ml phytohaemagglutinin (PHA). Two parallel cultures (duplicate cultures) for each individual were set up for selected concentrations (0.01–5-µg/ml) of fungicide. Thus, four cultures were set up for each dose of CP. Lymphocyte cultures were incubated in 5 % CO2 at 37 °C for 72 h. Cytochalasin-B at 6 µg/ml final concentration was added to each culture at 44 h and the cultures were harvested 72 h after initiation. CP was added to the lymphocyte culture 48 h after initiation. Thus, cells were treated with CP for 24 h. A positive control (1 µg/ml MMC) was established in parallel. At the end of the culture period, the cells were fixed. At this stage, cold 0.075 M KCl was used as a hypotonic solution and cold acetic acid/methanol (1:7 v/v) as a fixative. Slides were stained with 5 % Giemsa (double filtered) for microscopical analysis. The evaluation of slides was carried out in accordance with Fenech’s microscopic survey-criteria, at 1000× magnification (Fenech 2000). A total of 4000 bi-nucleated cells with well-preserved cytoplasm (1000 per replicate for each donor) were scored to determine the frequencies of MN for each concentration. The total number of micronuclei in the lymphocytes (MNL) and the total number of bi-nucleated cells with micronuclei (BNMN) were determined. Two thousand cells were also counted for each concentration to obtain CBPI. An assessment of cytotoxicity for each concentration was performed by reference to the CBPI in relation to the method described by Eastmond and Tucker (1989). The method was formulated as follows: CBPI = (MI + 2MII + 3MIII + 4MIV)/N. In this formula, MI, MII, MIII, and MIV represent the number of cells with 1, 2, 3, 4 nuclei, respectively. In addition, the percent cytostasis for each concentration was calculated by a formula recommended by Lorge, as follows: % cytostasis = 100 − 100 (CBPIT – 1)/(CBPIC – 1). CBPIT and CBPIC represent the CBPI in the fungicide-treated and control cultures, respectively (Lorge et al. 2008).
Statistical analysis
All results are presented as the mean (±SE) from two parallel experiments. Comparisons between concentrations of fungicides and control value were made using repeated-measure analysis of variance (ANOVA) with Dunnett’s multiple-comparison tests. Linear regression analysis was performed to show dose dependence. These tests were performed by means of GraphPad Prism 5 software for Windows. Significance was accepted at P < 0.05.
Results
Comet assay
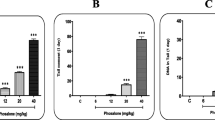
The result of the positive control (50 µM H2O2) for each experiment showed a statistically significant increase in DNA damage in comparison with control (P < 0.001). Cell viability after treatment in the comet assay was assessed by the Trypan-blue exclusion assay. The higher concentration of CP (5 µg/ml) caused ~10 % cell death. Figure 1 shows the results of primary DNA damage in isolated lymphocytes after treatment for 1 h with CP in the 0.01–5-µg/ml dose range. DNA damage significantly increased at 0.1, 1, 2.5 and 5 µg/ml of CP in comparison with DNA damage in untreated lymphocytes. Furthermore, linear regression analysis showed a statistically significant dose-dependent increase in primary DNA damage (R2 = 0.92, P < 0.01).

Effect of the fungicide Cabrio Plus on DNA damage (±SD) in lymphocytes from a female and a male human donor, after a 1-h exposure at 37 °C, measured by use of the comet assay; *P < 0.05; ***P < 0.001. AU: arbitrary unit. The arbitrary unit was calculated by use of the following equation: (% of cells in class 0)x0 + (% of cells in class 1)x1 + (% of cells in class 2)x2 + (% of cells in class 3)x3 + (% of cells in class 4)x4
Cytotoxicity and micronucleus formation
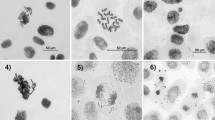
As recommended, at least three concentrations of CP with no more than √10-fold spacing between the concentrations were selected (Kirsch-Volders et al. 2000, 2003). In this test, cytotoxicity was assessed by counting the number of nucleated cells. The distribution of nucleated cells (mean ± SE) and the calculated cytotoxicity are presented in Table 1. The highest concentration of CP caused approximately 50 % toxicity. Furthermore, 25 µg/ml of CP induced ~70 % cytotoxicity and was not evaluated with the MN test. It can be seen that the CBPI decreased with increasing amounts of CP. The results of linear regression analysis showed a dose-dependent cytotoxicity in the peripheral blood lymphocytes (R2 = 0.74, P < 0.05). Statistically significant cell division delay caused by 0.1–5 µg/ml of CP. The percentages of MNL and BNMN obtained from two donors are given Table 2. The MN formation increased depending on CP concentrations for both donors. Combined results for two donors were given in Table 3. In the range of doses evaluated in the present study, CP increased the frequency of MN. 0.1–5 µg/ml of CP induced statistically significant MNL. Similarly, 1–5 µg/ml of CP significantly increased BNMN compared with cultures without CP. The positive control MMC induced statistically significant MN formation in comparison with negative control (P < 0.001).
Discussion
A number of in vitro studies with different test systems have been performed to show genotoxicity and/or cytotoxicity of different chemicals (Drozdz et al. 2011; Foldbjerg et al. 2011; Klarić et al. 2010; Viau et al. 2009; Wozniak and Blasiak 2004). The tests performed on cell cultures and/or in the animal model indicated that agrochemicals could significantly increase DNA damage. Furthermore, human bio-monitoring studies on the genotoxicity of pesticides, assessed with the micronucleus test, the sister-chromatid exchange (SCE) assay, the chromosomal aberration (CA) test, and the comet assay (Augusto et al. 1997; Coskun et al. 2011; Costa et al. 2006, 2007; Gomez-Arroyo et al. 2000; Martínez-Valenzuela et al. 2009; Paiva et al. 2011; Ündeğer and Başaran 2002), have shown that occupational exposure to such chemicals could induce a significant increase in genetic damage.
In this study, we investigated the genotoxic potential of the commercial fungicide Cabrio Plus by measuring micronucleus formation in whole blood after a 24-h treatment period. Primary DNA damage was evaluated in the in vitro culture of isolated human peripheral blood lymphocytes with the comet assay after a 1-h treatment period. To date, several studies have been performed to evaluate the genotoxic and/or cytotoxic potential of different commercial pesticide formulations by means of various test systems (Koller et al. 2012; Moretti et al. 2002; Ribas et al. 1997; Santovito et al. 2011; Undeger and Basaran 2005; Villarini et al. 1998). As far as is known, there are no reports related to DNA damage induced by the commercial formulation CP. Statistically significant increase in DNA damage was observed at 0.1, 1, 2.5 and 5 mg/ml concentrations of CP. Similarly, MN induction by CP concentrations (0.1–5 µg/ml) did reach a statistically significant level. In addition to genotoxic effect, the findings of the in vitro MN assay approach showed a clear cytotoxicity potential of CP. Apart from dose range (0.01–5 µg/ml), the concentration of 25 µg/ml of CP was also included, which induced highly toxic effects (~70 % cytotoxicity) on cell proliferation. With increasing concentrations, necrotic and apoptotic cells were observed, especially when the lymphocytes were treated with doses of 5 and 25 µg/ml. These cells were not evaluated further since the hypotonic treatment of whole blood may destroy the cytoplasm of necrotic and apoptotic cells (Fenech et al. 1999). The MN assay is a cytogenetic method requiring cell division in culture for the expression of the effect (Fenech et al. 1999). Thus, damage in cells at high concentrations may not be observed since they have undergone apoptosis and/or necrosis instead of completing nuclear division. In addition, higher cytotoxicity might result in mitotic delay and/or cell death, which may be responsible for the lower MN levels at high concentrations. For example, it has been suggested that the interaction of the insecticide nuvacron with cell-cycle kinetics causes lower MN formation at higher concentrations (Peitl et al. 1996).
CP contains metiram and pyraclostrobin as the active ingredients. Metiram is recognized as a probable carcinogen for humans by the US-EPA (2007). Detailed studies on genotoxicity in different test systems (gene mutation in bacteria and induction of chromosomal aberrations in rat bone-marrow) have indicated that metiram is not genotoxic. Although metiram induced sister chromatid exchange (SCE) in Chinese Hamster Ovary cells in the presence and absence of mouse microsomal activation, no significant induction of SCE was observed in the presence of rat microsomal activation (U.S. EPA 2007). Results of the potential for inducing MN are similar to previous studies of SCE induction.
As a member of the class of strobilurin fungicides, pyraclostrobin induces inhibition of mitochondrial respiration in fungi. It has been investigated for its genotoxic activity in vitro for mutagenicity in bacterial and mammalian cells, for chromosome damage (clastogenicity) and for unscheduled DNA synthesis, where it showed no effect. In contrast, pure pyraclostrobin (99.9 %) induced a significant increase in micronucleus frequency in human peripheral blood lymphocytes (Çayır et al. 2014). Similar genotoxic potential was reported by Bony et al. (2010) for azoxystrobin, which belongs to the same chemical group as pyraclostrobin. This agent induced micronucleus formation in erythrocytes and increased DNA damage, determined by use of the comet assay, in liver cells and spermatozoa of zebrafish (Bony et al. 2010). Another genotoxicity study revealed that azoxystrobin (0.5–1 μg/l) resulted in a 3–5-fold increase in DNA damage in erythrocytes of Phoxinus phoxinus, determined by comet assay, compared with unexposed controls (Bony et al. 2008).
Newly synthesized formulations described as pesticides have to pass several test systems before they can be used safely. An increase in DNA damage determined with the alkaline version of the comet assay and MN assay is indication of genotoxic potential of any pesticide. In the results obtained here, MN and comet formation revealed a dose-dependent genotoxic effect of CP. In the obtained results, MN and comet formation revealed a dose-dependent genotoxic effect of CP. In addition, according to dosage, CP was found to be cytotoxic on human peripheral blood lymphocytes. Both the comet assay and IVMNT were able to detect genetic damage induced by CP for different treatment time. We suggest that a more complete assessment of the effects of commercially formulated pesticides on human health be carried out to clarify the toxic potential of these mixtures.
References
Augusto LGD, Lieber SR, Ruiz MA, deSouza CA (1997) Micronucleus monitoring to assess human occupational exposure to organochlorides. Environ Mol Mutagen 29:46–52
Bony S, Gillet C, Bouchez A, Margoum C, Devaux A (2008) Genotoxic pressure of vineyard pesticides in fish: field and mesocosm surveys. Aquat Toxicol 89:197–203
Bony S, Gaillard I, Devaux A (2010) Genotoxicity assessment of two vineyard pesticides in zebrafish. Int J Environ Anal Chem 90:421–428. doi:10.1080/03067310903033659
Bøyum A (1976) Isolation of lymphocytes, granulocytes and macrophages. Scand J Immunol 5:9–15
Çayır A, Coskun M, Coskun M (2014) Micronuclei, nucleoplasmic bridges, and nuclear buds induced in human lymphocytes by the fungicide signum and its active ingredients (boscalid and pyraclostrobin). Environ Toxicol 29:723–732
Collins AR (2004) The comet assay for DNA damage and repair. Mol Biotechnol 26:249–261
Coskun M, Cayir A, Ozdemir O (2011) Frequencies of micronuclei (MNi), nucleoplasmic bridges (NPBs), and nuclear buds (NBUDs) in farmers exposed to pesticides in Canakkale, Turkey. Environ Int 37:93–96. doi:10.1016/j.envint.2010.08.002
Costa C, Teixeira JP, Silva S, Roma-Torres J, Coelho P, Gaspar J, Alves M, Laffon B, Rueff J, Mayan O (2006) Cytogenetic and molecular biomonitoring of a Portuguese population exposed to pesticides. Mutagenesis 21:343–350. doi:10.1093/mutage/ge1039
Costa C, Silva S, Coelho P, Roma-Torres J, Teixeira JP, Mayan O (2007) Micronucleus analysis in a Portuguese population exposed to pesticides: preliminary survey. Int J Hyg Environ Health 210:415–418. doi:10.1016/j.ijheh.2007.01.011
Cox C, Surgan M (2006) Unidentified inert ingredients in pesticides: implications for human and environmental health. Environ Health Perspect 114:1803
Dhawan A, Anderson D (2009) The comet assay in toxicology, vol 5. Royal Society of Chemistry, Cambridge
Dhawan A, Bajpayee MM, Pandey AK, Parmar D (2003) Protocol for the single cell gel electrophoresis/comet assay for rapid genotoxicity assessment. Sigma 1077:1
Drozdz K, Wysokinski D, Krupa R, Wozniak K (2011) Bisphenol A-glycidyl methacrylate induces a broad spectrum of DNA damage in human lymphocytes. Arch Toxicol 85:1453–1461
Eastmond DA, Tucker JD (1989) Identification of aneuploidy-inducing agents using cytokinesis-blocked human-lymphocytes and an antikinetochore antibody. Environ Mol Mutagen 13:34–43
Fenech M (2000) The in vitro micronucleus technique. Mutat Res Fundam Mol Mech Mutagen 455:81–95
Fenech M (2007) Cytokinesis-block micronucleus cytome assay. Nat Protoc 2:1084–1104. doi:10.1038/nprot.2007.77
Fenech M, Crott J, Turner J, Brown S (1999) Necrosis, apoptosis, cytostasis and DNA damage in human lymphocytes measured simultaneously within the cytokinesis-block micronucleus assay: description of the method and results for hydrogen peroxide. Mutagenesis 14:605–612
Foldbjerg R, Dang DA, Autrup H (2011) Cytotoxicity and genotoxicity of silver nanoparticles in the human lung cancer cell line, A549. Arch Toxicol 85:743–750
Gomez-Arroyo S, Díaz-Sánchez Y, Meneses-Pérez MA, Villalobos-Pietrini R, De León-Rodríguez J (2000) Cytogenetic biomonitoring in a Mexican floriculture worker group exposed to pesticides. Mutat Res Genet Toxicol Environ Mutagen 466:117–124
Kirsch-Volders M, Sofuni T, Aardema M, Albertini S, Eastmond D, Fenech M, Ishidate M, Lorge E, Norppa H, Surralles J, von der Hude W, Wakata A (2000) Report from the in vitro micronucleus assay working group. Environ Mol Mutagen 35:167–172
Kirsch-Volders M, Sofuni T, Aardema M, Albertini S, Eastmond D, Fenech M, Ishidate M, Kirchner S, Lorge E, Morita T, Norppa H, Surralles J, Vanhauwaert A, Wakata A (2003) Report from the in vitro micronucleus assay working group. Mutat Res/Genet Toxicol Environ Mutagen 564:97–100. doi:10.1016/j.mrgentox.2004.07.002
Klarić MŠ, Daraboš D, Rozgaj R, Kašuba V, Pepeljnjak S (2010) Beauvericin and ochratoxin A genotoxicity evaluated using the alkaline comet assay: single and combined genotoxic action. Arch Toxicol 84:641–650
Koller VJ, Fürhacker M, Nersesyan A, Mišík M, Eisenbauer M, Knasmueller S (2012) Cytotoxic and DNA-damaging properties of glyphosate and Roundup in human-derived buccal epithelial cells. Arch Toxicol 86:805–813
Lorge E, Hayashi M, Albertini S, Kirkland D (2008) Comparison of different methods for an accurate assessment of cytotoxicity in the in vitro micronucleus test I. Theoretical aspects. Mutat Res Genet Toxicol Environ Mutagen 655:1–3. doi:10.1016/j.mrgentox.2008.06.003
Martínez-Valenzuela C, Gómez-Arroyo S, Villalobos-Pietrini R, Waliszewski S, Calderón-Segura ME, Félix-Gastélum R, Álvarez-Torres A (2009) Genotoxic biomonitoring of agricultural workers exposed to pesticides in the north of Sinaloa State, Mexico. Environ Int 35:1155–1159
Moretti M, Marcarelli M, Villarini M, Fatigoni C, Scassellati-Sforzolini G, Pasquini R (2002) In vitro testing for genotoxicity of the herbicide terbutryn: cytogenetic and primary DNA damage. Toxicol In Vitro 16:81–88
Paiva JCG, Cabral IO, Soares BM, Sombra CML, Ferreira JRO, Moraes MO, Cavalcanti BC, Pessoa C (2011) Biomonitoring of rural workers exposed to a complex mixture of pesticides in the municipalities of Tianguá and Ubajara (Ceará state, Brazil): genotoxic and cytogenetic studies. Environ Mol Mutagen 52:492–501
Peitl P, SakamotoHojo ET, Colus IMD (1996) Genotoxic activity of the insecticide Nuvacron (Monocrotophos) detected by the micronucleus test in bone marrow erythrocytes of mice and in CHO cells. Braz J Genet 19:571–576
Ribas G, Surrallés J, Carbonell E, Xamena N, Creus A, Marcos R (1997) Genotoxic evaluation of the herbicide paraquat in cultured human lymphocytes. Teratog Carcinog Mutagen 17:339–347
Santovito A, Cervella P, Delpero M (2011) In vitro aneugenic effects of the fungicide thiabendazole evaluated in human lymphocytes by the micronucleus assay. Arch Toxicol 85:689–693
Singh NP, McCoy MT, Tice RR, Schneider EL (1988) A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 175:184–191
Tice R, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu J, Sasaki Y (2000) Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen 35:206–221
Undeger U, Basaran N (2005) Effects of pesticides on human peripheral lymphocytes in vitro: induction of DNA damage. Arch Toxicol 79:169–176. doi:10.1007/s00204-004-0616-6
Ündeğer ÜÜ, Başaran N (2002) Assessment of DNA damage in workers occupationally exposed to pesticide mixtures by the alkaline comet assay. Arch Toxicol 76:430–436
U.S. EPA (1997) Pesticide registration (PR) notice 97-6: use of term “inert” in the label ingredients statement. Accessed 6 Mar 2013
U.S. EPA (2005) What is a pesticide? http://www.epa.gov/pesticides/about/index.htm. Accessed 6 Mar 2013
U.S. EPA (2006) Terminology services. http://iaspub.epa.gov/sor_internet/registry/termreg/searchandretrieve/termsandacronyms/search.do. Accessed 6 Mar 2013
U.S. EPA (2007) Chemicals evaluated for carcinogenic potential. Health Effects Division, Office of Pesticide Programs, Office of Prevention, Pesticides, and Toxic Substances, Washington
Viau C, Guecheva TN, Sousa F, Pungartnik C, Brendel M, Saffi J, Henriques JAP (2009) SnCl 2-induced DNA damage and repair inhibition of MMS-caused lesions in V79 Chinese hamster fibroblasts. Arch Toxicol 83:769–775
Villarini M, Moretti M, Pasquini R, Scassellati-Sforzolini G, Fatigoni C, Marcarelli M, Monarca S, Rodríguez AV (1998) In vitro genotoxic effects of the insecticide deltamethrin in human peripheral blood leukocytes: DNA damage (‘comet’assay) in relation to the induction of sister-chromatid exchanges and micronuclei. Toxicology 130:129–139
Wozniak K, Blasiak J (2004) Vanadyl sulfate can differentially damage DNA in human lymphocytes and HeLa cells. Arch Toxicol 78:7–15
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Rights and permissions
About this article

Cite this article
Çayır, A., Coşkun, M. & Coşkun, M. Genotoxicity of commercial fungicide Cabrio Plus on human cell. Cytotechnology 68, 1697–1704 (2016). https://doi.org/10.1007/s10616-015-9919-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10616-015-9919-0