
One previously undescribed sesquiterpene analogue, M2 (1), together with five known ones, indole-3-carboxylic acid (2), methyl 4-hydroxycinnamate (3), ergosterol peroxide (4), 6β-hydroxystigmast-4-en-3-one (5), as well as betulonic acid (6), were isolated from the EtOAc extract of the culture broth of an actinomycete Streptomyces sp. M2. The structures of all isolates were elucidated on the basis of extensive analyses of spectroscopic data and comparison with literature data.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Actinobacteria, tiny microorganisms residing in the soil, are well-known for producing a diverse range of secondary metabolites showcasing various bioactivities, including antimicrobial and insecticidal properties. These compounds have proven to be crucial in developing new pharmaceuticals [1,2,3]. Among the Actinobacteria, Streptomyces species are particularly notable for their exceptional ability to produce antibiotics and other bioactive compounds. Strain M2, isolated from Chiayi’s Wetland, was identified as a Streptomyces species based on morphological characteristics and its 16S rDNA sequence analysis. Streptomyces encompasses a vast genus consisting of over 800 species, with many strains capable of generating biologically active secondary metabolites that hold significant medical importance. This specific strain thrives in neutral conditions and exhibits growth within a temperature range of 20 to 45°C. In dual culture assays, it demonstrated strong inhibitory activity against various pathogenic fungi, including Fusarium sp. LC8, Neopestalotiopsis sp. BCRC 35002, and Colletotrichum gloeosporioides BCRC 35178. Metabolic products derived from this strain displayed a diverse profile, as revealed by high-performance liquid chromatography (HPLC) fingerprinting analysis. Upon a thorough review of the existing literature, no references were found regarding the chemical and biological activity of the aforementioned Actinobacteria.
The scaled-up fermentation and extensive chromatographic separation of the EtOAc extract resulted in the isolation of one new metabolite, M2 (1), together with five known compounds, indole-3-carboxylic acid (2) [4], methyl 4-hydroxycinnamate (3) [5], ergosterol peroxide (4) [6], 6β-hydroxystigmast-4-en-3-one (5) [7], as well as betulonic acid (6) [8]. All of these compounds were found for the first time from the title Actinobacteria strain M2. Herein, we report the structural determination of the new compound.
Compound 1 was obtained as a colorless oil. The molecular formula was determined as C17H24O4 (four degrees of unsaturation) by HR-ESI-MS m/z 315.1573 [M + Na]+ (calcd for C17H24NaO4, 315.1572), which was in agreement with the 1H and 13C NMR data (Table 1). The UV spectrum absorption λmax (MeOH) at 226 nm, and a strong IR absorption at 1702 cm–1, as well as the observation of the featuring carbon resonances [δC 150.0 (C-11), 123.7 (C-12), and 172.3 (C-13)] in the 13C NMR spectrum (Table 1), revealed the presence of a conjugate carbonyl functionality in compound 1.
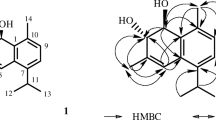
Its residual IR spectrum showed absorption bands for COOH (3400 cm–1), and one vinyl (1650 cm–1) functionalities. The 1H NMR spectrum (Table 1) of compound 2 showed the presence of one terminal double bond at δH 4.80/4.43, (1H, s, CH2-17), one (E)-11,12-double bond at δH 7.13 (1H, dd, J = 16.0, 11.2 Hz, H-11), 5.84 (1H, d, J = 16.0 Hz, H-12), and two Me moieties [δH 1.17 (3H, s, Me-19) and 0.89 (3H, s, Me-20)]. From 13C NMR data, it can be found that compound 1 was composed of 17 carbons. It is speculated that it was originally composed of 20 carbons, but the double bond was cleaved owing to the oxidation of the side chain. The C-20 labdane diterpene becomes a C-17 derivative. The presence of electron-withdrawing groups in proximity to the double-bond position causes a downfield shift in the signal of δH 7.13 (H-11), as observed. The HMBC analysis (Fig. 1) indicates a correlation between both H-11 and H-12 with C-13 (δC 172.3), which corresponds to a carboxyl group. The 13C NMR of C-19 shows another group of carboxyl groups at δC 183.8. The relative configuration of 1 was derived by a NOESY spectrum (Fig. 1), the relative configuration of which was based on a NOESY analyses. NOEs for Me-19/Me-20, and H-5/H-9 indicated that H-5/H-9 were on the same side of the molecular plane, tentatively assumed as α-orientation. The (E)-2-carboxyvinyl was β-oriented, which was further confirmed by the NOE H-11/H-20. Consequently, the relative configuration of C-4, 5, 9, and 10 was assigned as rel-(R,R,S,S) (Fig. 1). Thus, compound 1 was elucidated as M2, which was further confirmed by 13C NMR, COSY, HMBC, NOESY, and HSQC experiments.

Structure and key HMBC and NOESY correlations of compound 1.
Owing to the early development and research applications of Streptomyces species, our team has been isolating and collecting actinomycetes resources from various regions and environments in Taiwan over the years. As well as the common Streptomyces strains, we have also obtained numerous rare genera, including new species. Guided by the concept of “new species, new compounds,” we hope to discover unique compounds from these novel species in the future.
EXPERIMENTAL
General Procedures. TLC: silica gel 60 F254 precoated plates (Merck). Column chromatography (CC): silica gel 60 (230–400 mesh, Merck) and Spherical C18 100A Reversed Phase Silica Gel (RP-18) (particle size: 40–63 μm) (LiChroprep). Optical rotation: Jasco P-2000 polarimeter. IR spectra: Perkin-Elmer-2000 FT-IR spectrophotometer. 1H, 13C, and 2D NMR spectra: Varian-VNMRS-600 spectrometers; with Me4Si, as an internal standard. EI-MS: VG-Biotech Quatro-5022 mass spectrometer. ESI and HR-ESI-MS: Bruker APEX-II mass spectrometer. Reverse-phase HPLC (RP-HPLC): Hitachi model L-2310 pump equipped with a diode-array detector (model L-2455, Hitachi).
Microorganism and Fermentation. The actinomycete, Streptomyces sp. M2, was isolated from both the wetland of Houmei in Chiayi and soil samples from agricultural fields in Taiwan, by using HV agar, and was then incubated at 45°C for 7 days. These Actinobacteria were identified byMin Tseng and specimens were deposited at the Bioresource Collection and Research Center, Food Industry Research and Development Institute. The strain was maintained on oatmeal agar and the spores or mycelia suspension were harvested with 20% (v/v) glycerol and stored at –20°C. Liquid culture method was carried out in following six steps: 1. collect spores of the fungal strain; 2. inoculate the spores into separate TSB (Tryptic Soy Broth) and YG (Yeast Extract Glucose) culture media; 3. shake the cultures at 120 rpm for 48–72 h; 4. transfer the fungal broth to TSB and YG agar media for expansion culture; 5. shake the agar cultures at 120 rpm for 48–72 h. 6. The cultures are now ready for further use.
Fermented broth (1 L) was filtered to separate mycelium and culture broth. The culture broth was repeatedly extracted three times with EtOAc. The EtOAc layers were combined and dried to give a fraction soluble in EtOAc (≈ 4.4 g) and was chromatographed over silica gel (70–230 mesh), eluting with n-hexane and enriched with EtOAc to produce five fractions (1–5). Fraction 1 (170 mg) was chromatographed over silica gel and eluted with n-hexane–EtOAc (10:1→0:1) to afford three fractions (1-1–1-3). Fraction 1-2 (8.7 mg) was purified by preparative TLC (n-hexane–acetone, 2:1) to afford compounds 1 (7.9 mg) and 4 (2.8 mg). Fraction 2 (800 mg) was applied to silica gel (230–400 mesh, 24 g) and eluted with hexane–acetone (20:1) to give four fractions (2-1–2-4). Fraction 2-3 (32 mg) was chromatographed on a silica gel column (230–400 mesh, 1 g), eluting with n-hexane–acetone (10:1) to give six fractions (2-3-1–2-3-6). Fraction 2-3-1 was further purified by preparative TLC (n-hexane–acetone, 10:1) to afford compound 3 (1.4 mg). Fraction 2-3-5 (79 mg) was further purified by preparative TLC (n-hexane–acetone, 10:1) to afford compounds 2 (1.3 mg), 5 (2.8 mg), and 6 (2.6 mg).
M2 (1), oil; \({[{\upalpha }]}_{\mathrm{D}}^{25}\) –1.1° (c 0.08, CHCl3). UV (MeOH, λ, nm): 226 (2.18). IR (neat, ν, cm–1): 3400, 1702 (COOH), 1650 (C=C). For 1H (600 MHz, CDCl3) and 13C NMR (150 MHz, CDCl3), see Table 1. EI-MS m/z 293 [M + H]+; HR-ESI-MS m/z 315.1573 [M + Na]+ (calcd for C17H24NaO4, 315.1572).
References
H. Osada, Actinomycetol., 9, 254 (1995).
B. Mythili and D. M. P. Ayyappa, Res. J. Agric. Sci., 2, 104 (2011).
E. Kuster, The Actinobacteria, in: Soil Biology, eds. A. Burges & F. Raw, Academic Press, London, 1968, pp. 111–124.
H. Chiji, Y. Arakawa, S. Ueda, M. Kuroda, and M. Izawa, Phytochemistry, 25, 281 (1986).
Y. C. Chang, F. R. Chang, and Y. C. Wu, J. Chin. Chem. Soc, 47, 373 (2000).
C. E. Wu, K. Koike, T. Nikaido, K. Ishii, T. Ohmoto, and K. Ikeda, Chem. Pharm. Bull., 38, 2281 (1990).
Y. H. Kuo and P. H. Chu, J. Chin. Chem. Soc., 49, 269 (2002).
Ito, F. R. Chang, H. K. Wang, Y. K. Park, M. Ikegaki, N. Kilgore, and K. H. Lee, J. Nat. Prod., 64, 1278 (2001).
Acknowledgment
This work was supported by grants from the Ministry of Science and Technology. This work was kindly supported by the Ministry of Science and Technology, R.O.C. (MOST-108-2320-B-080-002-, MOST-109-2622-E-080-001-, MOST-110-2320-B-080-001-, and NSTC-111-2320-B-030-010-MY3). The authors thank Senior Technician Mrs. Chyi Jia Wang of the Center for Resources, Research, and Development (CRRD) of Kaohsiung Medical University for measuring the 2D NMR data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2024, pp. 739–741.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Cheng, MJ., Wu, MD., Chang, CL. et al. One Undescribed Sesquiterpene from Streptomyces sp.. Chem Nat Compd (2024). https://doi.org/10.1007/s10600-024-04462-9
Received:
Published:
DOI: https://doi.org/10.1007/s10600-024-04462-9