
A phytochemical investigation on the roots of Aconitum austroyunnanense led to the isolation of two previously undescribed diterpenoid alkaloids, austroyunnanines D and E (1 and 2). Their structures were determined by spectral methods such as 1D and 2D (1H–1H COSY, HMQC, NOESY, and HMBC) NMR spectroscopy, in addition to high-resolution mass spectrometry. The isolated compounds were tested in vitro for cytotoxic activity against three human gastric carcinoma cell lines. As a result, compound 1 exhibited some cytotoxicities against all the tested tumor cell lines with IC50 values less than 20.0 μM.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
The genus Aconitum (Ranunculaceae), which comprises 400 species, is naturally distributed throughout Northern Asia and North America [1, 2]. A widely used Chinese and Japanese herbal medicine, the tubers and roots of some species of the genus are mainly employed for the treatment of collapse, syncope, rheumatic fever, painful joints, gastroenteritis, diarrhea, oedema, bronchial asthma, various tumors, and some endocrinal disorders like irregular menstruation [3, 4]. The Aconitum alkaloids were structurally classified as C18-diterpenoid, C19-diterpenoid, and C20-diterpenoid alkaloids, which possess diverse bioactivities, including cytotoxic, antiplasmodial, anti-inammatory, antinociceptive, antioxidant, and tyrosinase inhibition activities [5,6,7,8]. We have reported the isolation of diterpenoid alkaloids from Aconitum austroyunnanense E. Pritz. ex Diels [9]. As an extension of a search for bioactive alkaloids, the present study was undertaken to systematically examine the roots of A. austroyunnanense affording two new diterpenoid alkaloids, austroyunnanines D and E (1 and 2). The structures of these compounds were elucidated mainly by NMR spectroscopic and mass spectroscopic methods; furthermore, the two alkaloids were evaluated in vitvo for their cytotoxic potential.
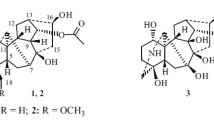
Compound 1 was obtained as a white amorphous powder, exhibiting a quasimolecular ion peak at m/z 482.2032 [M + H]+ in the high-resolution mass spectrometry, which corresponded to the molecular formula C23H31NO10 with 9 degrees of unsaturation. The IR spectrum showed the typical absorption assigned to OH (3444 cm–1), ester CO (1780 cm–1), and acetyl CO (1741 cm–1) functions. The 1H NMR spectrum of 1 exhibited proton signals for the presence of four methoxyl groups. Characteristic 13C NMR (Table 1) and HSQC spectra determined 23 skeleton carbons in 1, including five methylenes (one oxygenated), eight methines (three oxygenated) and six quaternary carbons (two carbonyl groups) and as well as four methoxys (δ 53.5, 55.0, 58.3, and 59.3). Inspection of its characteristic NMR data revealed that 1 possessed a rearrangedtype skeleton in C18-diterpenoid alkaloids similar to kusnezosine A [10]. The four methoxyl groups were attributed to C-1, C-6, C-15, and C-18 based on the HMBC correlations from 1-OCH3 (δ 3.22) to C-1 (δ 80.6), 6-OCH3 (δ 3.35) to C-6 (δ 82.0.), from 15-OCH3 (δ 3.85) to C-15 (δ 173.6), and from 18-OCH3 (δ 3.30) to C-18 (δ 80.3), respectively. The HMBC cross-peaks of H-17 (δ 3.56) to C-5 (δ 49.4), C-6, and C-8 (δ 74.2) indicated that C-17 was connected with C-7.

A carbonyl groups were located at C-8 based on the HMBC cross-peaks from H-7 (δ 1.98, d, J = 6.6 Hz) and H-9 (δ 2.97, m) to C-8 (δ 74.2). A nitrone group was placed between C-17 and C-19, which was confirmed by the deshielding shifts of H-17, H-19 (δ 6.95), C-17 (δ 71.2), and C-19 (δ 145.2). The presence of a double bond between the N-atom and C-19 was verified by the HMBC correlations between the olefinic H-19 and C-4, C-5 and C-17. The long-range HMBC correlations between H-12 (δ 1.94 and 2.91, each, m) and H-14 (δ 8.10, d, J = 6.6 Hz) to C-16 (δ 177.6) suggested the ketone group of C-16. The stereochemistry of compound 1 was deduced from the NOESY experiment. The NOESY correlations between H-10 and H-1, H-5, H-7, H-9, H-14, and 13-OH elucidated that they were all β-oriented. Thus, the structure of compound 1 was structurally elucidated and named austroyunnanine D.
Compound 2 was isolated as a white amorphous powder with the molecular formula C25H37NO7, as determined by positive-ion HR-ESI-MS, showing an [M + H]+ ion at m/z 464.2643. The IR spectrum showed the typical absorption assigned to OH (3423 cm–1) and acetyl CO (1738 cm–1). In the 1H NMR spectrum, the signals of an olefinic H atom at δ 5.47 (d, J = 6.4 Hz, H-7), two OMe groups at δ 3.22 and 3.34, and one N-ethyl [δ 2.38 and 2.58 (each, m, -NCH2Me) and 0.95 (3H, t, J = 7.0 Hz, -NCH2Me] were observed. The 13C NMR and DEPT spectrum displayed 25 carbon signals, which were identified with the aid of the DEPT experiment as one acetyl group, three methyls (two oxygenated), seven methylenes (one olefinic and one oxygenated), nine methines (one olefinic and four oxygenated), and four quaternary carbons. The spectroscopic data revealed that compound 2 was an aconitine-type C19-diterpenoid alkaloid possessing a structure similar to the known acotarine D [11]. The mere 13C NMR difference of the downfield shift of signals to δ 49.2 to 84.6 for C-10 in 2, relative to those of acotarine D, indicated that C-10 was linked with one hydroxyl group. The cross-peaks of H-6 and H-7 with C-8, of H-6 with C-7, and of H-7 with C-10 in the HMBC spectrum suggested that the C=C bond should be positioned between C-7 and C-8. Furthermore, the cross-peaks between H-6 and H-5/H-7, H-12 and H-13, H-16 and H-13/H-15 observed in the 1H–1H COSY spectrum supported the above conclusions. The NOESY correlations of H-1 with H-5, and H-9, of H-5 with H-18, and of H-13 with H-9 and MeO-16 indicated that they were all β-oriented. The relative configuration of compound 2 was established by the NOESY spectrum as being the same as acotarine D. Based on the above interpretation, the structure of compound 2 was substantiated and named austroyunnanine E.
The diterpenoid alkaloids obtained from genus Aconitum have been reported to show strong antioxidant activities [12, 13], which agrees well with our results. The isolated compounds were in vitro evaluated for their cytotoxic potential by using the revised MTT method. The results showed that compound 1 exhibited some cytotoxicity against three human gastric carcinoma cell lines (MGC-803, BGC-823, and SGC-7901) with IC50 values of 17.6, 14.3, 15.8 μM, respectively (IC50 values of doxorubicin: 2.3, 3.1, 3.7 μM), while no cytotoxicity was detected for compound 2 (IC50 ≥ 50 μM).
Experimental
General Procedures. Optical rotations: Perkin-Elmer 341 polarimeter. UV Spectra: Hewlett-Packard-8452A diode-array spectrophotometer. IR Spectra: Nicolet Magna FT-IR 750 spectrophotometer in cm–1. 1H and 13C NMR spectra: Bruker AM-600 spectrometer. HR-ESI-MS spectra: Micromass LC-MS/MS mass spectrometer. Column chromatography (CC): silica gel (200–300 mesh, 10–40 μm; Qingdao Marine Chemical Factory, Qingdao, China), Sephadex LH-20 (Amersham Pharmacia Biotech, Sweden). Thin-layer chromatography (TLC): silica gel GF254 (10–40 μm; Qingdao Marine Chemical Factory, Qingdao, China). All solvents were distilled before use. Preparative HPLC: Varian SD1 instrument with a 320 single-wave detector on C-18 columns (250 × 10 mm, 5 μm, Waters; 220 × 25 mm, 10 μm, Merck, respectively).
Plant Material. The roots of Aconitum austroyunnanense were collected at the Dali of Yunan Province of China in June of 2022. A specimen (AA20220601), identified by one of the authors (X. Mao), was deposited in the Herbarium of the College of Biological Resources and Food Engineering, Qujing Normal University, Qujing, Yunnan, China.
Extraction and Isolation. The dried roots of A. austroyunnanense (10.0 kg) were powdered and percolated with 0.05 M HCl (50 L) at room temperature. Wet resin (type: 001 × 7, dry weight 30 kg) was added to the percolate, followed by repeated washing on a suction filter with deionized H2O. The air-dried resin was then alkalized with 10% aq. NH4OH (3.0 L) and continuously extracted with Et2O (5.0 L), and evaporated to yellowish amorphous powder. The yellowish amorphous powder was suspended in H2O (l L) and then partitioned with CHCl3 (8.0 L), and evaporated to render the total crude alkaloids as a yellowish amorphous powder. The crude alkaloids (98.0 g) were chromatographed over SiO2 column (2 kg, 200–300 mesh) and eluted with petroleum ether (PE)–acetone–Et2NH (from 10:1:1 to 5:1:1) to provide five fractions (F1–F5). Fraction F3 (4.2 g) was chromatographed on a SiO2 column eluting with CHCl3–MeOH, 90:10 to afford three fractions F3a (817 mg), F3b (976 mg), and F3c (948 mg). Fraction F3b (842 mg) was chromatographed on preparative HPLC (MeOH–H2O, from 60% to 70%, 240 nm, 220 × 25 mm, 10 μm, Merck), yielding 2 (66 mg, retention time: 14.6 min). Fraction F4 (2.9 g) was chromatographed on a SiO2 column eluting with CHCl3–MeOH (85:15) to afford three fractions, F4a–F4c. Fraction F4c (201 mg) was further separated by preparative HPLC (MeOH–H2O, from 55% to 65%, 240 nm, 220 × 25 mm, 10 μm, Merck) to afford 1 (69 mg, retention time: 14.9 min).
Austroyunnanine D (1), white amorphous powder, [α]25D –16.87° (c 0.10, CHCl3). IR (KBr, νmax, cm–1): 3444, 3365, 2962, 2922, 2854, 1780, 1741, 1659, 1466, 1260, 1095, 803. UV (MeOH, λmax, nm) (log ε): 195 (3.72). 1H and 13C NMR data, see Table 1. HR-ESI-MS m/z 482.2032 [M + H]+ (calcd for C23H32NO10, 482.2026).
Austroyunnanine E (2), white amorphous powder, [α]25D –11.64° (c 0.10, CHCl3). IR (KBr, νmax, cm–1): 3423, 2930, 2822, 1738, 1637, 1454, 1385, 1365, 1244, 1206, 1095, 1053. UV (MeOH, λmax, nm) (log ε): 196 (3.92). 1H and 13C NMR data, see Table 1. HR-ESI-MS m/z 464.2643 [M + H]+ (calcd for C25H38NO7, 464.2648).
Cytotoxicity Assay in vitro. The isolated compounds (1 and 2) were subjected to cytotoxic evaluation against three human gastric carcinoma cell lines (MGC-803, BGC-823, and SGC-7901) by employing the revised MTT method [14]. Doxorubicin was used as the positive control. All tumor cell lines were cultured on RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin in 25 cm2 culture flasks at 37°C in a humidified atmosphere with 5% CO2. For the cytotoxicity tests, cells in the exponential growth stage were harvested from culture by trypsin digestion and centrifuged at 180 × g for 3 min, then resuspended in fresh medium at a cell density of 5 × 104 cells per mL. The cell suspension was dispensed into a 96-well microplate at 100 μL per well, incubated in a humidified atmosphere with 5% CO2 at 37°C for 24 h, and then treated with the compounds dissolved in DMSO at various concentrations (0, 1, 10, 100 μM). After 48 h of treatment, 50 μL of 1 mg/mL MTT solution was added to each well, and further incubated for 4 h. The cells in each well were then solubilized with DMSO (100 μL for each well) and the optical density (OD) was recorded at 570 nm. All cell lines were purchased from the Cell Bank of Shanghai Institute of Biochemistry & Cell Biology, Chinese Academy of Sciences.
References
H. Niitsu, Y. Fujita, S. Fujita, R. Kumagai, M. Takamiya, Y. Aoki, and K. Dewa, Forensic Sci. Int., 227, 111 (2013).
J. Singhuber, M. Zhu, S. Prinz, and B. Kopp, J. Ethnopharmacol., 126, 18 (2009).
A. Bello-Ramirez, J. Buendia-Orozco, and A. Nava-Ocampo, Fund. Clin. Pharmacol., 17, 575 (2003).
T. Yamamoto, K. Yuyama, K. Nakamura, T. Kato, and H. Yamamoto, Eur. J. Pharmacol., 397, 25 (2000).
J. Li, W. Ren, X. J. Huang, D. J. Zou, and X. Hu, Brain Res. Bull., 97, 81 (2013).
T. Kiss, P. Orvos, S. Bansaghi, P. Forgo, N. Jedlinszki, L. Talosi, J. Hohmann, and D. Csupor, Fitoterapia, 90, 85 (2013).
M. Liang, S. C. Li, B. Shen, J. P. Cai, C. Li, Z. Y. Wang, X. G. Li, J. Gao, H. Y. Huang, X. Y. Zhang, and J. Y. Li, Carbohyd. Polym., 88, 973 (2012).
D. K. Zhao, H. L. Ai, S. H. Zi, L. M. Zhang, S. C. Yang, H. C. Guo, Y. Shen, Y. P. Chen, and J. J. Chen, Fitoterapia, 91, 280 (2013).
J. Hu, Q. Wu, Q. Li, T. Lv, T. F. Peng, S. Yin, and H. Z. Jin, J. Asian Nat. Prod. Res., 25, 132 (2023).
Y. Z. Li, L. L. Qin, F. Gao, L. H. Shan, and X. L. Zhou, Fitoterapia, 144, 104738 (2020).
Y. Si, X. Ding, T. A. Adelakuna, Y. Zhang, and X. J. Hao, Fitoterapia, 147, 717 (2020).
Y. P. Fu, Y. F. Z, F. Y. Lei, and H. Wangensteen, J. Ethnopharmacol., 291, 115148 (2022).
H. L. Tao, X. F. g Liu, R. M. Tian, Y. Liu, Y. Zeng, X. L. Meng, and Y. Zhang, J. Ethnopharmacol., 301, 115726 (2023).
J. Hu, Y. Song, X. Mao, Z. Wang, and Q. J. Zhao, J. Funct. Foods, 20, 1 (2016).
Acknowledgment
The above research was made possible by a grant from the joint special fund project of the Applied Basic Research for Local University in Yunnan Province (2019FH001 (-033)), Special funds for the Reform and Development of the Local University supported by the Central Finance (PL201707230369), and the National Undergraduate Training Programs for Innovation and Entrepreneurship (201910684029).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2024, pp. 415–417.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Hu, J., Li, G., Xu, F. et al. Cytotoxic Diterpenoid Alkaloids from Aconitum austroyunnanense. Chem Nat Compd 60, 472–475 (2024). https://doi.org/10.1007/s10600-024-04354-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-024-04354-y