
Two new coumarin derivatives, 4-(3,5-dihydroxyphenyl)-3,5,7-trihydroxy-2H-chromen-2-one (1) and 4-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-(((2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)-2H-chromen-2-one (2), along with five known compounds, were isolated for the first time from ethyl acetate fraction of Sterculia colorata. Structures of isolated compounds were characterized using spectroscopic techniques, such as EI-MS, 1H NMR, and 13C NMR. Compounds 1 and 2 showed significant immunomodulatory (IC50 = 11.42 ± 0.62 μg/mL and 10.98 ± 0.24 μg/mL) and anticancer activities (IC50 = 2.85 ± 0.18 μg/mL and 2.62 ± 0.15 μg/mL), respectively.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Sterculia colorata Roxb., known as Jungli chalghouza in Urdu, can be found in Myanmar, Sri Lanka, Thailand, Southern Yunnan, Vietnam, Bangladesh, Pakistan, and India [1]. It belongs to the genus Sterculia of the family Sterculiaceae, usually found in hill tracts [2]. Sterculia is famous for a variety of medicinal uses and has been scientifically proven to show diverse pharmacological effects, including anti-inflammatory, analgesic, antipyretic, immunomodulatory, anticancer, insecticidal, larvicidal, leishmanicidal, anthelmintic, cytotoxic, fibrinolytic, antibacterial, antiglycation, antioxidant, laxative, antidiarrheal, antihyperglycemic, anticonvulsant, and antidepressant properties [3,4,5,6,7,8,9,10].
Traditionally, Sterculia colorata was used for the treatment of jaundice and cholera. Sterculia colorata possesses antioxidant and antidiarrheal activities in the leaf and bark extracts [1]. Various types of phytochemicals are also present in the plant seeds, such as malvalate, sterculate, oleate, palmitate, linoleate, stearate, and myristate. [11].
In this study, the major chemical constituents of Sterculia colorata and their bioactivities were investigated. As a result, two new compounds, 4-(3,5-dihydroxyphenyl)-3,5,7-trihydroxy-2H-chromen-2-one (1) and 4-(3,4-dihydroxyphenyl)-5,7- dihydroxy-3-(((2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)-2H-chromen-2-one (2) (Fig. 1), together with five known compounds, rutin (3), myricetin (4), lupeol (5), β-sitosterol (6), and β-sitosterol-D-glucoside (7), were isolated from Sterculia colorata. Their structures were elucidated by extensive analysis of their spectral data. Herein, we report the isolation and structural elucidation of the new compounds 1 and 2 as well as their immunomodulatory and anticancer activities.

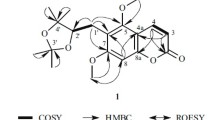
Structures and key COSY and HMBC correlations of compounds 1and 2.
Compound 1 was obtained as an yellowish amorphous powder. It has a molecular formula of C15H10O7, which was established on the basis of HR-ESI-MS at m/z 302.2421. The UV (methanol) absorption maxima occur at 222, 298, and 330 nm. The IR spectrum at 3430 cm–1 region has a broad band matching to phenolic OH stretching. Owing to carbonyl and aromatic stretching there are bands at 1710 and 1503 cm–1.
The 1H NMR spectrum (Table 1) showed signals at δ 7.82 (2H, s), 6.82 (t, J = 2.4 Hz), 6.32 (s), and 6.25 (s). The 13C NMR and HSQC data (Table 1) revealed 15 carbon signals, including nine signals from ten quaternary carbons (δ 159.2, 158.8, 122.6, 160.3, 158.9, 157.0, 104.9, 140.2, and 150.6 (2C)) and four signals from five methine carbons (δ 110.6 (2C), 102.4, 100.4, and 97.6).
The 2D NMR technique was applied for further structure confirmation of compound 1. The HMBC correlations of compound 1 from H-6 to C-5, C-7, C-8, and C-10 as well as from H-8 to C-7 and C-9 confirmed the position of the 5,7-dihydroxyphenyl moiety. Furthermore, the HMBC correlations from H-2′ to C-4, C-1′, C-3′, and C-6′ as well as H-4′ to C-3′ and C-5′, whereas H-6′ to C-4 and C-5′ confirmed the position of a 3,5-dihydroxyphenyl group at C-4. The planar structure of compound 1 was further confirmed by the 1H–1H COSY and HMBC spectra (Fig. 1). The chemical shifts and other data of 1 have not matched to known compounds in literature and databases, and hence it was concluded to be a new compound.
Thus, the structure of compound 1 was confirmed as shown in Fig. 1, and it was named as 4-(3,5-dihydroxyphenyl)-3,5,7- trihydroxy-2H-chromen-2-one (1).
Compound 2 was obtained as an yellow amorphous powder with a molecular formula of C21H20O12 as determined by analysis of positive EI-MS (m/z 465.3802 [M + H]+). The absorption characteristic for flavonoids was shown through IR spectrum at 1433 cm–1 (double bonds of aromatic rings) and 3415 (OH). The stretching of C–H appeared at 2918 cm–1. The conjugated system was indicated by UV absorption bands at λmax 213, 260, and 360 nm.
The 1H NMR (Table 1) showed a meta coupled doublet at δ 7.73 (1H, d, J = 2.0 Hz, H-2′) with H-6′. The H-6′ also showed meta coupling with H-2′ at δ 7.57 (dd, J = 8.4, 2.0 Hz), whereas H-5′ at δ 6.86 (1H, d, J = 8.4 Hz) showed an ortho coupling with H-6′ of aromatic ring B. Similarly, H-8 and H-6 appeared at δ 6.20 (1H, d, J = 2.0 Hz) and 6.28 (1H, d, J = 2.0 Hz), respectively.
The ring C contains a glucose moiety substituent showing the anomeric proton at δ 5.15 (1H, d, J = 6.8 Hz). The H-2′′ and H-3′′ of glucose moiety appeared at δ 3.89 (1H, dd, J = 6.8, 1.6 Hz) and 3.81 (1H, m). Similarly, the H-4′′, H-5′′, and H-6′′ exhibited at δ 3.63 (1H, dd, J = 8.4, 2.8 Hz), 3.46 (1H, m), and 3.82 (1H, m), 3.42 (1H, m), respectively. The 13C NMR (BB and DEPT) showed 21 carbon signals, including one methylene, 10 methine, and 10 quaternary carbon signals. C-5 and C-7, the OH substituted carbon atoms of aromatic ring A, exhibited at δ 161.2 and 164.1. Similarly, C-3′ and C-4′, the OH substituted carbon atoms of aromatic ring B, exhibited at δ 144.8 and 148.5. C-9 and C-10 are quaternary carbon atoms. They exhibited at δ 156.2 and 104.0, whereas C-6 and C-8 methine carbon atoms appeared at δ 98.7 and 93.5. The aromatic ring B methines at C-2′, C-5′, and C-6′ exhibited at δ 115.2, 116.5, and 121.6, respectively. The glucose moiety present in this compound possesses anomeric carbon C-1′′ exhibiting at δ 100.9, whereas OH-substituted carbon atoms C-2′′, C-3′′, and C-4′′ exhibited at δ 72.3, 76.5, and 70.0, respectively. Methine C-5′′ and methylene C-6′′ exhibited at δ 77.6 and δ 61.0, respectively. The H-2′ shows the HMBC correlations with the aromatic ring B carbon atoms C-1′ and C-3′, as well as with C-4 of ring C. The H-6′ shows correlations with C-4 of ring C, whereas it also shows connectivity with C-1′ and C-4′ of aromatic ring B. The H-5′ shows correlations with C-1′, C-3′, and C-4′ of aromatic ring B. The H-8 of aromatic ring A shows correlations with C-6 methine carbon, C-7 (OH substituted carbon), and C-9 and C-10 quaternary carbon atoms. The H-6 position was confirmed through HMBC correlations. Similarly, the glucose moiety anomeric proton shows correlations with C-2′′ and C-3′′ carbon atoms. The chemical shifts and other data of 2 have not matched to known compounds in literature and databases, and hence it was concluded to be a new compound. Thus, the structure of 2 was confirmed as shown in Fig. 1 and named as 4-(3,4-dihydroxyphenyl)-5,7-dihydroxy-3-(((2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2- yl)oxy)-2H-chromen-2-one (2). It is isolated for the first time from any source and its complete NMR data have also been reported for the first time.
By comparison of their spectral data, as well as physicochemical properties with those reported in the literature, five known compounds were identified as rutin (3) [12], myricetin (4) [13], lupeol (5) [14], β-sitosterol (6) [15], and β-sitosterol-D-glucoside (7) [16].
Subsequently, isolated compounds 1 and 2 were subjected to immunomodulatory and anticancer activity assays (Table 2). Compounds 1 and 2 showed significant immunomodulatory activity with IC50 values of 11.42 ± 0.62 μg/mL and 10.98 ± 0.24 μg/mL, respectively, compared with ibuprofen (standard), IC50 value of 10.84 ± 0.92 μg/mL. Compounds 1 and 2 were also investigated for anticancer activity against PC-3 cell line. Compounds 1 and 2 showed highly significant results, IC50 values of 2.85 ± 0.18 μg/mL and 2.62 ± 0.15 μg/mL, respectively, compared with doxorubicin (standard), IC50 value of 2.48 ± 0.28 μg/mL.
EXPERIMENTAL
General. Melting points were recorded by using Gallenkamp apparatus and optical rotations were determined by using JASCO DIP-360 digital polarimeter (JASCO, Tokyo, Japan). UV spectra were recorded in methanol by using Shimadzu spectrophotometer (Shimadzu, Columbia, ML, USA) spectra and IR data were collected in chloroform on a JASCO A-320 IR Spectrophotometer (JASCO). EI-MS spectra were scanned on a JEOL-JMS HX-110 mass spectrometer (JEOL, Peabody, MA, USA). 1H NMR spectra were recorded on a Bruker, AM-300, AM-400, and AM-500 NMR spectrometers (Bruker, Faellanden, Switzerland) with TMS as the internal standard. 13C NMR spectra were measured on the same instruments. The DEPT (distortionless enhancement by polarization transfer) experiments were recorded at different degrees, i.e., 45°, 90°, and 135° for determining CH, CH2, and CH3 functionalities. The signals of these spectra were subtracted from that of the BB (broad band) 13C NMR for determination of quaternary carbons. Silica gel PF254 (Merck, Darmstadt, Germany) and pre-coated silica gel F254 (Merck) were used for column chromatography and thin-layer chromatography (TLC), respectively.
Plant Material. Sterculia colorata is the plant used in this research work and was collected from Lahore, Punjab, Pakistan in 2019 and was identified by a plant taxonomist in the Department of Botany, University of Peshawar, Peshawar, Pakistan. Voucher specimens were submitted in the Herbarium of the Department of Botany [17].
Extraction and Isolation. The shade-dried whole plant (14 kg) was extracted in methanol (90%) at ambient temperature. The whole filtrate was concentrated using a rotary evaporator (Zhengzhou Great Wall Scientific Industrial and Trade Co., Zhengzhou, China) to give 1.15 kg of extract [18]. This extract was separated in different organic solvents on gradient polarity basis to attain different fractions, such as n-hexane (70.5 g), chloroform (109.5 g), ethyl acetate (204.3 g), n-butanol (342.3 g), and aqueous (323.4 g). Ethyl acetate fraction (25.0 g) was added to column chromatography using 300 g silica gel (Merck, Darmstadt, Germany) as a stationary phase. The column was eluted with n-hexane–chloroform, chloroform–methanol, and methanol on increasing polarity to obtain subfractions [19]. Analysis was performed on TLC plates. According to the TLC results, four subfractions were obtained. Subfractions S.C2 and S.C3 were subjected to column chromatography for further extraction. Subfraction S.C2 was rechromatographed with n-hexane–EtOAc (6:4) to yield compounds 1 (32 mg), 3 (rutin, 68 mg), 4 (myricetin, 59 mg), 5 (lupeol, 10 mg), and 6 (β-sitosterol, 12 mg). Subfraction S.C3 was purified using n-hexane–EtOAc (5:5) to generate compounds 2 (20 mg) and 7 (β-sitosterol-D-glucoside, 23 mg). Compounds 3–7 were isolated from this plant for the first time.
4-(3,5-Dihydroxyphenyl)-3,5,7-trihydroxy-2H-chromen-2-one (1). Yellowish amorphous powder (MeOH), \({[\mathrm{\alpha }]}_{\mathrm{D}}^{25}\) +17.4° (MeOH). UV (MeOH, λmax, nm): 222, 298, 330. IR (KBr, νmax, cm–1): 3430 (hydroxyl), 1710 (C=O), and 1503 (aromatic). 1H and 13C NMR (400 MHz, CD3OD) spectroscopic data, see Table 1. HR-ESI-MS m/z 302.2421 [M]+ (calcd for C15H10O7, 302.2319).
4-(3,4-Dihydroxyphenyl)-5,7-dihydroxy-3-(((2R,3S,4R,5R,6S)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)-2H-chromen-2-one (2). Yellowish amorphous powder (MeOH), \({[\mathrm{\alpha }]}_{\mathrm{D}}^{26}\) –75° (MeOH). UV (MeOH, λmax, nm): 213, 260, 360. IR (KBr, νmax, cm–1): 3415 (hydroxyl), 2918 (C–H), and 1433 (aromatic). 1H and 13C NMR (400 MHz, CD3OD) spectroscopic data, see Table 1. HR-ESI-MS m/z 464.3802 [M]+ (calcd for C21H20O12, 464.3695).
Bioassays. Immunomodulatory Assay. Luminol-enhanced chemiluminescence assay was performed using standard protocol [20]. Briefly, whole blood (diluted 1:200) neutrophils (1 × 107) and polymorphonuclear leukocytes (1 × 106) were suspended in Hank′s balance salt solution (HBSS) with calcium and magnesium and incubated with 50 μL of test compound (prepared using methanol as solvent) concentrations (1.6 to 50 μg/mL) for 30 min. To each well, 50 μL (20 mg/mL) zymosan (Sigma Chemical Co. USA), followed by 50 μL (7 × 10s M) luminal (G-9382 Sigma Chemical Co.), and then HBSS were added to adjust the final volume to 0.2 mL. HBSS was used as a control. Chemiluminescence′s peaks were recorded with a luminometer (Luminoskan RS Lab, Finland).
Anticancer (PC-3 Cell Line) Activity. Anticancer activity was recorded in 96-well microplates by MTT assay. Human prostate cancer cells (PC-3) were cultured in Dulbecco′s Modified Eagle′s Medium along with 5% FBS (Fetal bovine serum), 100 μg/mL streptomycin and 100 IU/mL penicillin in a 75-cm2 flask and kept in a 5% CO2 incubator at 37°C. Exponentially growing cells were harvested, counted with a hemocytometer, and diluted using the medium. Cell culture was prepared with the concentration of 1 × 105 cells/mL and introduced (100 μL/well) into 96-well plates. Medium was removed after incubation and fresh medium (200 μL) was added with concentrations of test samples (1–30 μM). The test sample was prepared using methanol as the solvent. After 48 h, 200 μL MTT (0.5 mg/mL) was added to each well and incubated further for 4 h. 100 μL DMSO was added to each well. By measuring the absorbance (570 nm), MTT reduction was calculated using a microplate reader. The cytotoxicity was measured as a concentration causing 50% growth inhibition (IC50) for PC-3 cells [21].
The percentage inhibition was determined by using the following formula:
Statistical Analysis. Both assays were carried out in triplicate, and the results obtained were reported as mean of IC50 and its standard deviation using GraphPad Prism software (version 9.0 for Windows, GraphPad Software, San Diego, CA, USA).
References
S. Tabassum, Bachelor Thesis, BRAC University, 93 (2018).
L. Sonawane, P. Sonawane, and A. Gujar, Life Sci., 17, 246 (2020).
A. Shukla, K. Desai, and N. Modi, Future J. Pharm. Sci., 6, 1 (2020).
M. M. El-Sherei, A. Y. Ragheb, M. E. S. Kassem, M. M. Marzouk, S. A. Mosharrafa, and N. A. M. Saleh, Asian Pac. J. Trop. Dis., 6, 492 (2016).
A. Das, J. J. Jawed, M. C. Das, P. Sandhu, U. C. De, B. Dinda, Y. Akhter, and S. Bhattacharjee, Int. J. Antimicrob. Agents, 50, 512 (2017).
E. Wilson, M. Chacha, and J. Omolo, J. Pharmacogn. Phytochem., 3, 125 (2014).
M. B. Oppong, L. Yang, P. O. Banahene, F. Shi-Ming, and Q. Feng, Chin. J. Nat. Med., 16, 721 (2018).
F. Rabbi, A. Zada, A. Nisar, F. Khan, A. Rahim, M. Salman, A. Iqbal, A. Ullah, and M. Sohail, J. Anim. Plant Sci., 31, 296 (2021).
S. R. Lamia, S. Afra, M. R. Hoque, S. Paul, and S. Sumon, Int. J. Res. Pharm. Pharm. Sci., 5, 1 (2020).
F. Rabbi, A. Zada, A. Adhikari, A. Nisar, and S. K. Khalil, J. Asian Nat. Prod. Res., 23, 899 (2021).
C. Daulatabad and R. Ankalgi, J. Am. Oil Chem. Soc., 59, 439 (1982).
H. Soni, J. Malik, A. P. Yadav, and B. Yadav, Eur. J. Biomed. Pharm. Sci., 5, 484 (2018).
C. Wen, D. Wang, X. Li, T. Huang, C. Huang, and K. Hu, J. Pharm. Biomed. Anal., 150, 144 (2018).
H. H. Shwe, K. K. Win, T. T. Moe, A. A. Myint, and T. Win, IEEE-SEM, 7, 140 (2019).
J. Sawale, A. Yadav, R. Thagele, B. Patel, A. Saxena, A. Bagre, R. Gonekar, A. Bhardwaj, N. Jain, and R. Sagar, RKDF Univ. J. Sci. Eng., 1, 69 (2018).
S. Desai, P. Tatke, T. Mane, and S. Gabhe, Food Chem., 345, 128717 (2021).
F. A. Alhumaydhi, A. S. Aljohani, U. Rashid, Z. A. Shah, A. Rauf, N. Muhammad, Y. S. Al-Awthan, and O. S. Bahattab, ACS Omega, 6, 996 (2021).
F. Hadi, A. Rahim, and F. Rabbi, Pharm. Chem. J., 55, 929 (2021).
N. Pournaghi, F. Khalighi-Sigaroodi, E. Safari, and R. Hajiaghaee, Iran. J. Pharm. Res., 20, 274 (2021).
A. O. Oladimeji, I. A. Oladosu, A. Jabeen, A. Faheem, M. A. Mesaik, and M. S. Ali, Pharm. Biol., 55, 2240 (2017).
I. S. Gade, Nat. Prod. Res., 36, 4085 (2022).
Acknowledgment
We express our gratitude to the Department of Pharmacy, University of Peshawar, Pakistan for providing the research facility. Further, we would also like to extend our gratitude to the HEJ Research Institute of Chemistry, the International Centre for Chemical and Biological Sciences, University of Karachi, Karachi, Pakistan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2023, pp. 371–374.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rabbi, F., Nisar, A. Two New Coumarins with Immunomodulatory and Anticancer Activities from Sterculia colorata. Chem Nat Compd 59, 436–440 (2023). https://doi.org/10.1007/s10600-023-04018-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-023-04018-3