
One novel naphthopyrone-derived metabolite, named bennepyrone A (1), was isolated from a marine crinoid Comanthus bennetti. The structure of the compound was elucidated on the basis of its spectroscopic data and comparison with those of comaparvin. Compound 1 possesses a new carbon skeleton that is derived from the naphthopyrone skeleton.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Studies of the chemical constituents of genus Comanthus have led to the isolation of various naphthopyrones [1,2,3,4,5,6,7,8]. Our previous chemical investigation on Comanthus bennetti had led to the isolation of one marine-derived compound comaparvin [9]. Moreover, this natural product comaparvin has been shown to have significantly anti-inflammatory and analgesic effects [9]. In this paper, we further report the isolation of one novel naphthopyrone-derived metabolite, bennepyrone A (1). Furthermore, it is noteworthy to mention that compound 1 has a new carbon skeleton that is derived from a naphthopyrone skeleton.
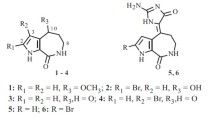
Bennepyron A (1) was isolated as a yellow powder. The HR-ESI-MS spectrum of 1 exhibited a molecular ion peak at m/z 395.1105 [M + Na]+, with the molecular formula C20H20O7, implying eleven degrees of unsaturation. IR absorptions were observed at 3420 and 1712 cm–1, suggesting the presence of hydroxyl and carbonyl groups in 1. The 13C NMR and DEPT spectroscopic data of 1 (Table 1) showed the presence of 20 carbons (Table 1): three methyls (including one methoxyl at 57.1), three sp3 methylenes, three sp2 methines, and eleven quaternary carbons (including one oxygenated carbon at δ 77.6, one α,β-unsaturated keto carbonyl at δ 198.5, and one ketone carbonyl at δ 205.0). From the 1H NMR spectrum of 1, the resonances of three olefinic protons (δ 7.27, s; 6.29, s; 5.57, s) and three methyls (δ 3.97, s; 2.12, s; 1.03, t, J = 7.0 Hz) were observed.

The structure and all of the 1H and 13C chemical shifts of 1 were elucidated by 2D NMR spectroscopic analysis, in particular 1H–1H COSY and HMBC experiments (Fig. 1). From the 1H–1H COSY spectral analysis, we established one partial structure of consecutive proton spin systems extending from H2-1′ to H3-3′. The following key HMBC correlations permitted connection of the carbon skeleton: H2-1′ to C-2 and C-3; H-3 to C-2 and C-4a; H-6 to C-4a, C-5, and C-7; H-9 to C-7, C-8, C-10, and C-10a. Moreover, comparison of the NMR data (Table 1) of 1 with comaparvin (2) confirmed that both compounds have the same partial structure from B and C rings (Fig. 1). Furthermore, one acetonyl moiety attached at C-7 was confirmed by the HMBC correlations from H2-11 to C-6a, C-7, C-8, and C-12, and from H3-13 to C-12. On the basis of the above findings and other detailed HMBC correlations, the relative structure of 1 was established.

Selected 1H–1H COSY and HMBC correlations of 1.
Experimental
General. Optical rotation values were measured with a Jasco P-1010 digital polarimeter. IR spectra were recorded on a Varian Digilab FTS 1000 Fourier transform infrared spectrophotometer. NMR spectra were recorded on a Varian Unity INOVA 500 FT-NMR instrument at 500 MHz for 1H NMR and 125 MHz for 13C NMR, respectively. ESI-MS were obtained with a Bruker APEX II mass spectrometer. Gravity column chromatography was performed on silica gel (230–400 mesh, Merck). TLC was carried out on precoated Kieselgel 60 F254 (0.2 mm, Merck), and spots were visualized by spraying with 10% H2SO4 solution followed by heating. High-performance liquid chromatography (HPLC) was performed using a system comprised of a Hitachi L-7100 pump and a Rheodyne 7725 injection port. A preparative normal phase column (Hibar 250 × 21.2 mm, Supelco, silica gel 60, 5 μm) was used for HPLC.
Animal Material. C. bennetti (Specimen No. MC2008-1) was collected by scuba divers at a depth of 10–15 m from coral reefs off the coast of Pingtung, Taiwan, in July 2008. A voucher specimen was deposited in the National Museum of Marine Biology and Aquarium, Taiwan.
Extraction and Isolation. The C. bennetti was frozen for storage and then freeze dried. The freeze-dried material (785 g) was minced and extracted exhaustively with CH2Cl2–MeOH (1:1) (4 × 2 L). The residue was triturated with n-hexane first to afford the n-hexane soluble layer, and with CH2Cl2, and then with n-butanol. The n-hexane soluble layer was evaporated under reduced pressure to afford a residue (47.6 g), which was subjected to column chromatography on silica gel using n-hexane and n-hexane–CH2Cl2 mixtures of increasing polarity, and finally pure acetone, to yield 32 fractions. Fraction 25 was subjected to normal phase HPLC using n-hexane–acetone (4:1) as eluent to afford 1 (3.0 mg).
Bennepyron A (1), yellow powder. \( {\left[\upalpha \right]}_{\mathrm{D}}^{24} \) +5.6° (c 0.08, CHCl3). UV (MeOH, λmax, nm) (log ε): 225 (3.87), 270 (3.84), 322 (3.51), 358 (3.48). IR (neat, νmax, cm–1): 3420, 2959, 2928, 1712, 1658, 1600, 1433, 1265. 13C and 1H NMR data, see Table 1. ESI-MS m/z 395 [M + Na]+; HR-ESI-MS m/z 395.1105 [M + Na]+ (calcd for C20H20O7Na, 395.1107).
References
Y. Chovolou, S. S. Ebada, W. Watjen, and P. Proksch, Eur. J. Pharmacol., 657, 26 (2011).
H. R. Bokesch, L. K. Cartner, R. W. Fuller, J. A. Wilson, C. J. Henrich, J. A. Kelley, K. R. Gustafson, J. B. McMahon, and T. C. McKee, Bioorg. Med. Chem. Lett., 20, 3848 (2010).
F. Folmer, W. T. Harrison, J. N. Tabudravu, M. Jaspars, W. Aalbersberg, K. Feussner, A. D. Wright, M. Dicato, and M. Diederich, J. Nat. Prod., 71, 106 (2008).
M. Sakurai, J. Kohno, K. Yamamoto, T. Okuda, M. Nishio, K. Kawano, and T. Ohnuki, J. Antibiot. (Tokyo), 55, 685 (2002).
Y. Sakuma, J. I. Tanaka, and T. Higa, Aust. J. Chem., 40, 1613 (1987).
K. A. Francesconi, Aust. J. Chem., 33, 2781 (1980).
G. L. Bartolin, T. R. Erdman, and P. J. Scheuer, Tetrahedron, 29, 3699 (1973).
I. R. Smith and M. D. Sutherland, Aust. J. Chem., 24, 1287 (1971).
L.-C. Chen, Y.-Y. Lin, Y.-H. Jean, Y. Lu, W.-F. Chen, S.-N. Yang, H.-M. D. Wang, I.-Y. Jang, I.-M. Chen, J.-H. Su, P.-J. Sung, J.-H. Sheu, and Z.-H. Wen, Molecules, 19, 14667 (2014).
Acknowledgment
This study was supported in part by a grant from the Ministry of Science and Technology, Taiwan and in part by a grant from the Antai Medical Care Cooperation, Antai Tian-Sheng Memorial Hospital Research Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 1, January–February, 2021, pp. 76–77.
Rights and permissions
About this article

Cite this article
Lin, TC., Lin, YY., Wu, YJ. et al. A Novel Naphthopyrone-Derived Metabolite from the Marine Crinoid Comanthus bennetti. Chem Nat Compd 57, 91–93 (2021). https://doi.org/10.1007/s10600-021-03290-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-021-03290-5