
A new diterpene, dysokusone G (1), was isolated from the stems of Dysoxylum lukii Merr., along with twelve known compounds, which were reported for the first time from this plant. The structures were elucidated using spectroscopic methods and by comparison with published NMR spectroscopic data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Dysoxylum lukii Merr. (Meliaceae family) is widely distributed in Guangxi, Guangdong, and Yunnan Provinces in China [1]. The previous studies on the constituents of D. lukii led to ergostane steroids, triterpenoids, limonoids, and diterpenes, some of which possessed cytotoxic, antimicrobial, and anti-PTP1B activities [2,3,4]. We studied the stems of D. lukii. A new diterpene 1, together with 12 known ones, was obtained.
Compound 1 was isolated as a colorless oil, with the molecular formula C20H30O2 provided by HR-ESI-MS (m/z 325.2139 [M + Na]+) and the 13C NMR spectrum, indicating six degrees of unsaturation. The IR absorptions of 1 indicated the presence of a carbonyl (1661, 1617 cm–1). In the 1H NMR spectrum (Table 1), the signals of two vinylic methyl groups [δH 2.13 (3H, s), 1.88 (3H, s)], a tertiary methyl group [δH 0.90 (3H, s)], and two secondary methyl groups [δH 0.92 (6H, d, J = 6.6 Hz)] were observed. The 13C NMR (Table 1) spectrum and HMQC experiments showed five methyls (δC 17.6, 22.7, 16.8, and 21.8), five methylenes (δC 54.3, 28.0, 39.8, 25.1, and 53.6), three methines (δC 49.5, 47.6, and 25.5), one quaternary carbon (δC 37.4), two carbonyl carbons (δC 198.8 and 201.6), and two double bonds (δC 127.0, 162.3, 160.4, and 122.7). The characteristics of the 1H and 13C NMR data suggested that 1 was a prenyleudesmane-type diterpene similar to dysokusone F [4], but H-5 of 1 was axial and assigned to be β-oriented, according to the coupling constant of H-5 with H-6 (J = 11.7 Hz) [5] and the NOESY correlation H-5 (δH 2.39) with H-7 (δH 2.12).
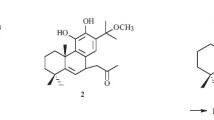
The known compounds were identified by comparison of their NMR and MS spectroscopic data with those reported. Compounds 2–13 were characterized as polylauioid H (2) [6], 2-oxoneoclerod-3-en-15-ol (3) [7], roseostachenone (4) [8], roseostachone (5) [8], 2-oxo-kolavenic acid methyl ester (6) [9], ent-3β,4β-epoxyclerod-13E-en-15-ol (7) [10], 14-ent-halimadien-3β,13S-diol (8) [11], (3α,4β)-3-O-acetylclerod-14-ene-4,13-diol (9) [12], α-hydroxytuberculosinol (10) [13], (+)-13-epi-2α-hydroxykolavelool (11) [14], agbanindiol A (12) [15], and (3α,4β,13E)-neoclerod-13-ene 3,4,15-triol (13) [11].

EXPERIMENTAL
General Experimental Procedures. Optical rotations were measured on a Perkin Elmer 341 polarimeter (Massachusetts, USA). IR spectra were recorded on a Perkin Elmer FT-IR spectrometer (KBr disc) (Massachusetts, USA). NMR spectra were recorded with a Bruker Avance 600 NMR spectrometer (Fallanden, Switzerland), and chemical shifts (δ) were expressed in ppm with reference to the solvent signals. MS data were obtained on a Bruker Daltonics Bio-TOF-Q mass spectrometer (Karlsruhe, Germany). Silica gel (200–300 mesh) and silica gel GF254 precoated plate (Qingdao Haiyang Chemical Inc., Qingdao, China) were used for column chromatography and TLC, respectively. Solvents used for extraction and isolation were distilled prior to use. HPLC separation was performed using an instrument consisting of a Perkin Elmer 600 controller (Massachusetts, USA), a Perkin Elmer 600 pump (Massachusetts, USA), and a Perkin Elmer 600 UV detector (Massachusetts, USA) (208 nm) with a Kromasil ( 250 × ∅ 10 mm) preparative column and a flow rate of 3 mL/min.
Plant Material. The stems of Dysoxylum lukii Merr. were collected in August 2008 from Xishuangbanna District, Yunnan Province, P. R. China. The sample was identified by Prof. Jing-Yuan Cui at Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences (CAS). A voucher specimen (No. 08–52) was deposited in the Herbarium of Chengdu Institute of Biology, CAS.
Extraction and Isolation. The air-dried stems of D. lukii Merr. (4.0 kg) were powdered and percolated with 95% EtOH (3 × 15 L, each 6 days) at room temperature. The solvents were evaporated under reduced pressure. The remaining gum (186 g) was suspended in water (2 L) and fractionated with ethyl acetate (3 × 4 L). The ethyl acetate fraction (150 g gum) was divided into eleven fractions (Frs. A–K) over a silica gel column (200–300 mesh, 30 × ∅ 10 cm) eluted gradiently with petroleum ether–acetone (10:1–1:1) based on TLC analysis. Fraction B (5 g) was separated using HPLC (silica 60 column) using hexane–isopropanol (50:1) as solvent to yield 1 (23 mg). Fraction C (10 g) was separated by HPLC (C18 column) with CH3OH–H2O (3:2) as solvent to give 4 (5 g), 5 (3 g), and Fr. C-1 (0.5 g). Fraction C-1 was further separated by HPLC with a silica 60 column [hexane–isopropanol (30:1)] to yield 6 (10 mg) and 7 (10 mg). Fraction D (5 g) was separated over a Sephadex LH-20 column (170 × ∅ 3 cm, CHCl3–MeOH, 1:1) and then recrystallized from MeOH to give 8 (4 g). Compound 9 (6 g) and Fr. E-1 were obtained from the separation of Fr. E (9 g) over a Sephadex LH-20 column (170 × ∅ 3 cm, CHCl3–MeOH, 1:1). Fraction E-1 (1 g) was separated to afford 3 (17 mg) and 10 (10 mg) with HPLC (silica 60 column) using hexane–isopropanol (20:1) as solvent. Fraction G was separated by HPLC with a silica 60 column (hexane–isopropanol, 20:1) to obtain 11 (7 mg), 12 (8 mg), and 2 (4 mg). Following the same separation procedure as that of Fr. G, 13 (9 mg) was obtained from Fr. J (5 g).
Dysokusone G (1). Colorless oil; \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –80.0° (c 0.1, CHCl3). IR (KBr, νmax, cm–1): 2938, 2871, 1661, 1617, 1437, 1377. HR-ESI-MS m/z 325.2139 [M + Na]+ (calcd for C20H30O2Na, 325.2143). For 1H and 13C NMR, see Table 1.
References
Editorial Committee of Flora of China, Flora Republicae Popularis Sinicae [in Chinese], Science Press, Beijing, 1997, Vol. 43, 93 pp.
J. Hu, Y. Song, X. D. Shi, X. Mao, J. G. Chen, and L. Zhu, Helv. Chim. Acta, 96 (12), 2245 (2013).
J. Hu, X. Wang, and X. D. Shi, Eur. J. Org. Chem., 2012 (9), 1857 (2012).
P. Z. Zhang, Y. Lin, F. Wang, D. M. Fang, and G. L. Zhang, Phytochem. Lett., 29 (1), 53 (2019).
J. Gu, S. Y. Qian, Y. L. Zhao, G. G. Cheng, D. B. Hu, B. H. Zhang, Y. Li, Y. P. Liu, and X. D. Luo, Tetrahedron, 70 (6), 1375 (2014).
Z. X. Yu, C. J. Zheng, G. Y. Chen, R. L. Rong, X. M. Zhou, Z. G. Niu, X. B. Li, C. R. Han, and X. P. Song, J. Nat. Prod., 82 (1), 27 (2019).
L. M. X. Lopes and V. S. Bolzani, Phytochemistry, 27 (7), 2265 (1988).
C. Fazio, S. Passamanti, M. P. Paternostro, and F. Piozzi, Phytochemistry, 31 (2), 3147 (1992).
C. M. Hasan, T. M. Healey, and P. G. Waterman, Phytochemistry, 21 (6), 1365 (1982).
F. Nagashima, H. Tanaka, Y. Kan, S. Huneck, and Y. Asakawa, Phytochemistry, 40 (1), 209 (1995).
S. M. Yang, S. H. Wu, X. D. Qin, X. D. Luo, and D. G. Wu, Helv. Chim. Acta, 87 (5), 1279 (2004).
K. M. Meragelman, L. A. Espinar, V. E. Sosa, M. L. Uriburu, and F. J. R. Dela, Phytochemistry, 41 (2), 499 (1996).
C. Nakano and T. Hoshino, Chembiochem., 10 (15), 2413 (2009).
M. D. Bomm, J. Zukerman-Schpector, and L. M. X. Lopes, Phytochemistry, 50 (3), 455 (1999).
D. E. U. Ekong and J. I. Okogun, J. Chem. Soc. C, 1969 (16), 2153 (1969).
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (Nos. 31760092 and 21562027) the China Scholarship Council and the Scientific Foundation of Double First-Class Discipline of Jiangxi University of TCM (JXSYLXK-ZHYAO110).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 3, May–June, 2020, pp. 394–395.
Rights and permissions
About this article

Cite this article
Zhang, Pz., Zhang, Ym. A New Diterpene from Dysoxylum lukii. Chem Nat Compd 56, 455–457 (2020). https://doi.org/10.1007/s10600-020-03061-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-020-03061-8