
Conjugates of betulin and betulinic and betulonic acids with 2-aminoethane- and N-methyl-2-aminoethanesulfonic acids were synthesized for the first time and were interesting as potential biologically active compounds. Experiments in vitro in MDCK cell culture using the MTT assay found that betulin and betulinic-acid derivatives with aminoethanesulfonic acid bound to triterpene C-3 or C-28 through an ester linker were less toxic than the native compounds.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Lupane-type pentacyclic triterpenoids are interesting as platforms for developing new drugs with various biological activities [1,2,3,4]. In continuation of research on the modification of lupane triterpenoids, we developed approached to conjugates containing aminoalkanesulfonic acids. The endogenous metabolite taurine (2-aminoethanesulfonic acid), its derivatives, and natural compounds containing taurine possess broad spectra of biological activity [5,6,7] including antiviral [8,9,10,11,12], antibiotic [13], and antitumor [14, 15]. Introduction of taurine or its homologs into lupane-type pentacyclic triterpenoids would expand the spectrum of biologically active compounds in this series and provide new information about the structure–activity (biological) relationship.
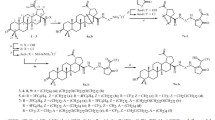
Herein, conjugates containing C-28/C-3 2-aminoethane- or N-methyl-2-aminoethanesulfonic acid (N-methyltaurine) as the ammonium or sodium salt or the free aminoethanesulfonic acids bound to the triterpene skeleton via an ester linker were synthesized from betulin (1) chloroacetates, betulonic acid (2) bromoalkyl esters, and betulinic acid (3a) methyl chloroacetates or benzyl esters. Experiments in vitro assessed the toxicity of the synthesized compounds in MDCK cell culture (canine kidney cell line).
Starting chloroacetates 4 and 5a,b were obtained from 1 or 3b,c and chloroacetic acid using the carbodiimide method. Betulonic acid 2-bromoethyl and 3-bromopropyl esters 6a,b were synthesized via O-alkylation of acid 2 with the appropriate dibromoalkane (DMF, K2CO3). Acid 2 was prepared by a modified literature method [16] using sequential oxidation of 1 by pyridinium chlorochromate (PCC) (2 h) and treatment of the obtained betulone aldehyde with NaClO2–NaH2PO4 in the presence of 2-methyl-2-butene for 15 min with an aldehyde–NaClO2–NaH2PO4 ratio of 1:6:6. The yield of acid 2 after purification through the potassium salt was 75%. Acid 3a and methyl 3b and benzyl 3c esters were synthesized by the reported methods [17,18,19]. N-Methyltaurine was prepared by N-alkylation of methylamine with sodium 2-bromoethanesulfonate in 73% yield [20].
Conjugates of taurine 7 and 8 or N-methyltaurine 9 and 10a,b were synthesized by N-alkylation of the tetrabutylammonium salt of taurine [21] or N-methyltaurine chloroacetates 4 and 5a or 5b in DMF in the presence of K2CO3 at 95°C.

Betulonic acid derivatives with sodium N-methyl-2-aminoethanesulfonate 11a and 11b were prepared by reacting bromoalkyl esters 6a and 6b with the sodium salt of N-methyltaurine [22] in the presence of K2CO3 in DMF at 95°C for 50 h.
The yields of conjugates 7–9, 10a,b, and 11a,b were 64–90%. The structures of these compounds were established using PMR and 13C NMR spectroscopy and mass spectrometry. PMR and 13C NMR spectra of conjugates 8, 9, 10b, and 11a were fully assigned using 2D experiments (Table 1).
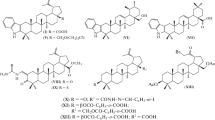
Toxicity in vitro of 1, 2, 3a, 7–9, 10a, and 11a. Assessment of the toxicity of the compounds was an important step in preclinical studies of their biological activity. The toxicity of synthesized 7–9, 10a, and 11a was screened in vitro using MDCK cell culture and the MTT assay. The median cytotoxic dose (CTD50, dose leading to the death of 50% of the cells) and minimum toxic dose (MTD, dose causing toxic effects to appear in the cells) were determined. The MTD became an independent important toxicity characteristic if the CTD50 value exceeded the upper limit of the tested concentrations. According to this criterion, conjugates 7, 9, and 10a were slightly toxic compounds (Table 2). A criterion reflecting the rate of toxicity increase with increasing concentration (CTD50/MTD) served as a guide for comparing the toxicity of compounds with similar CTD50 values, e.g., 8 (CTD50 30 μg/mL) and 11a (CTD50 37.5 μg/mL). According to this criterion, 11a (CTD50/MTD = 1.3; for 8, 4.8) was the most toxic compound.
The toxicity results for 7–9 and 10a showed that adding aminoethanesulfonic acids through ester linkers to triterpene C-3 or C-28 reduced considerably the toxicity of the conjugates as compared with native precursors 1 and 3a (Table 2, compounds 1, 7, 9, 3a, 8, 10a). The exception was betulonic acid derivative 11a, the toxicity of which was the same as acid 2 itself (CTD50/MTD = 1.5 and 1.6, respectively). An analysis of the toxicities of betulin and betulinic acid derivatives with the same substituents (pairs 7,8 and 9,10a) (Table 2) or derivatives within each series 7,9 and 8,11a with different substituents showed that the toxicity depended on both the triterpenoid structure and the fine differences in the substituents.
Thus, methods for preparing conjugates of betulin and betulinic and betulonic acids with aminoethanesulfonic acids that were interesting as potential biologically active compounds were developed. They were also intermediates for synthesizing various sulfonamide and peptide derivatives of lupane-type pentacyclic triterpenoids. The in vitro experiments provided seminal information on the effects of aminoethanesulfonic acid substituents on the toxicity of these compounds that was valuable for planning further syntheses of biologically active compounds based on aminoethanesulfonic acids.
Experimental
IR spectra were taken in mineral oil on a Shimadzu Prestige-21 IR spectrophotometer. PMR and 13C NMR spectra were recorded on a Bruker Avance III pulsed spectrometer at operating frequencies 500.13 MHz for 1H and 125.47 MHz for 13C using a Z-gradient PABBO probe at 298 K or a Bruker AM-300 spectrometer at operating frequencies 300.13 MHz for 1H and 75.47 MHz for 13C. Chemical shifts in PMR and 13C NMR spectra were given in ppm relative to solvent (CDCl3) resonances (δH 7.27 and δC 77.1) or TMS internal standard. 2D spectra were recorded using standard multi-pulse sequences of the Bruker Avance III spectrometer software. Mass spectra were obtained using electrospray ionization (ESI) or chemical ionization at atmospheric pressure (APCI) on an LCMS-2010EV liquid chromatograph-mass spectrometer (Shimadzu) (sample injection via syringe, 0.1 mL/min, MeCN–H2O eluent, 95:5) in positive- and negative-ion modes at capillary potential 4.5 and –3.5 kV. The APCI interface temperature was 250°C, heater 200°C, vaporizer 230°C. The spray-gas (N2) flow rate was 1.5 and 2.5 L/min for ESI and APCI, respectively. Rotation angles were measured on a PerkinElmer 341C instrument. Column chromatography used SiO2 (PTSKh-AF-A, Imid, Krasnodar, Russia). Melting points were determined on a Kofler apparatus. Betulin (1) was isolated from birch bark by the literature method [23]. Spectral characteristics (PMR and 13C NMR) of betulinic acid 3a and its esters 3b and 3c that were prepared according to the literature [17,18,19] agreed with those reported. Commercial 2-aminoethanesulfonic acid was used. DMF was stored over MgSO4 and vacuum distilled under Ar from 4Å molecular sieves. CH2Cl2 was distilled from CaH2. K2CO3 was calcined before the reactions.
3-Oxolup-20(29)-en-28-oic Acid (betulonic acid) (2). A solution of 1 (5.00 g, 11.29 mmol) in CH2Cl2 (500 mL) was treated with PCC (12.17 g, 56.47 mmol), stirred for 2 h, diluted with Et2O (500 mL), stirred for 15 min, and filtered through a layer of Al2O3. The filtrate was concentrated. The residue was purified by flash chromatography over SiO2 [C6H6–methyl-tert-butylether (MTBE), 4:1] to afford betulonic aldehyde (4.20 g, 85%) (PMR and 13C NMR spectra agreed with the literature [16]). A solution of the aldehyde (4.20 g, 9.57 mmol) in t-BuOH (200 mL) was treated with 2-methyl-2-butene (4 mL, 37.75 mmol) and simultaneously dropwise with NaClO2 (5.20 g, 57.44 mmol) in H2O (22 mL) and NaH2PO4 (6.89 g, 57.44 mmol) in H2O (21 mL). The reaction mixture was stirred for 15 min, diluted with H2O (200 mL), and extracted with CHCl3. The organic layer was separated, washed with H2O, dried over Na2SO4, and evaporated. The resulting residue was dissolved in C6H6 (150 mL), treated with KOH (0.58 g, 10.36 mmol, 15% solution in H2O), and refluxed for 1 h. The resulting precipitate of the potassium salt of betulonic acid was filtered off and dissolved in a mixture of MTBE (200 mL) and HCl (10%, 50 mL). The organic layer was separated, washed with H2O, dried over Na2SO4, and evaporated to afford 2 (3.26 g, 75%). The PMR and 13C NMR spectra agreed with the literature data [16].
3 β -Hydroxylup-20(29)-en-28-yl 2-Chloroacetate (4). A solution of 1 (3.00 g, 6.78 mmol) and Et3N (2.83 mL, 20.33 mmol) in CH2Cl2 (300 mL) was stirred, treated dropwise with a solution of chloroacetic chloride (1.3 mL, 16.26 mmol) in CH2Cl2 (50 mL), stirred at 20°C for 30 min, washed with HCl (10%, 100 mL) and H2O, dried over Na2SO4, and evaporated.
The residue was chromatographed over SiO2 (C6H6–MTBE, 8:1) to afford chloroacetate 4 (2.18 g, 63%), mp 152–154°C (lit. [24]: 154–156°C). IR spectrum (ν, cm–1): 1660, 1740, 3325. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.67 (1H, d, J = 8.9, H-5), 0.76 (3H, s, CH3-24), 0.82 (3H, s, CH3-25), 0.96 (3H, s, CH3-23), 0.98 (3H, s, CH3-26), 1.03 (3H, s, CH3-27), 1.68 (3H, s, CH3-30), 2.43 (1H, td, J = 10.5, 5.5, H-19), 3.18 (1H, dd, J = 11.0, 5.3, H-3), 3.86 (2H, s, OC(O)CH2), 3.94, 4.37 (each 1H, d, J = 11.0, H-28), 4.60, 4.70 (each1H, br.s, H-29) (only characteristic resonances are given). The 13C NMR spectrum agreed with the literature data [24].
Preparation of 5a and 5b. A solution of ester 3b or 3c (1.06 mmol) in CH2Cl2 (60 mL) was treated under Ar with chloroacetic chloride (2.13 mmol) and DMAP (2.13 mmol), stirred for 5 min, treated with DCC (2.13 mmol), and stirred for 20 min. The precipitate of N,N-dicyclohexylurea was filtered off. The organic layer was washed with HCl (10%, 20 mL) and H2O, dried over Na2SO4, and evaporated. The residue was purified by flash chromatography over SiO2 (C6H6–MTBE, 4:1).
Methyl Ester of 3 β -(2-Chloroacetoxy)lup-20(29)-en-28-oic Acid (5a). Yield 0.54 g (94%), amorph., \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +4° (c 0.15, CHCl3). IR spectrum (ν, cm–1): 1650, 1734. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.68 (1H, d, J = 10.0, H-5), 0.84 (3H, s, CH3-24), 0.85 (3H, s, CH3-25), 0.86 (3H, s, CH3-23), 0.91 (3H, s, CH3-27), 0.95 (3H, s, CH3-26), 1.68 (3H, s, CH3-30), 1.87 (2H, m, H-21, 22), 2.19 (1H, m, H-13), 2.22 (1H, m, H-16), 3.00 (1H, td, J = 11.0, 4.5, H-19), 3.66 (3H, s, C(O)OCH3), 4.05 (2H, s, CH2Cl), 4.56 (1H, dd, J = 10.0, 6.0, H-3), 4.60, 4.73 (each 1H, s, H-29). 13C NMR spectrum (75.47 MHz, CDCl3, δ, ppm): 14.64 (C-27), 16.13 (C-25), 16.40 (C-26), 15.92 (C-24), 18.10 (C-6), 19.31 (C-30), 20.86 (C-11), 23.52 (C-2), 25.41 (C-12), 27.88 (C-23), 29.63 (C-15), 30.55 (C-21), 32.12 (C-16), 34.17 (C-7), 36.92 (C-22), 37.06 (C-10), 37.97 (C-4), 38.18 (C-13), 38.28 (C-1), 40.65 (C-8), 41.23 (CH2Cl), 42.37 (C-14), 49.41 (C-19), 51.26 (C(O)OCH3), 46.95 (C-18), 50.40 (C-9), 55.35 (C-5), 56.51 (C-17), 83.35 (C-3), 109.62 (C-29), 150.52 (C-20), 167.14 (C=O), 176.64 (C-28). Mass spectrum (ESI), m/z 453 [M+H-ClCH2CO2H]+ (calcd for C33H51ClO4, 546.5).
Benzyl Ester of 3 β -(2-Chloroacetoxy)lup-20(29)-en-28-oic Acid (5b). Yield 0.61 g (92%), mp 57–59°C, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –9° (c 0.22, CHCl3). IR spectrum (ν, cm–1): 700, 1650, 1730, 1762. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.76 (3H, s, CH3-24), 0.83 (3H, s, CH3-25), 0.84 (3H, s, CH3-26), 0.85 (3H, s, CH3-23), 0.94 (3H, s, CH3-27), 1.68 (3H, s, CH3-30), 1.88 (2H, m, H-21, 22), 2.18 (1H, td, J = 11.0, 5.4, H-13), 2.28 (1H, d, J = 11.8, H-16), 3.02 (1H, td, J = 10.5, 4.2, H-19), 4.02, 4.05 (each1H, d, J = 18.5, CH2Cl), 4.55 (1H, dd, J = 10.5, 4.3, H-3), 4.60, 4.72 (each 1H, s, H-29), 5.08, 5.18 (each 1H, d, J = 19.6, CH 2Ph), 7.36 (5H, m, Ph). 13C NMR spectrum (75.47 MHz, CDCl3, δ, ppm): 14.64 (C-27), 15.83 (C-25), 16.19 (C-26), 16.43 (C-24), 18.14 (C-6), 19.35 (C-30), 20.90 (C-11), 23.56 (C-2), 25.46 (C-12), 27.94 (C-23), 29.55 (C-15), 30.55 (C-21), 32.10 (C-16), 34.18 (C-7), 36.94 (C-22), 37.07 (C-10), 38.00 (C-4), 38.15 (C-13), 38.31 (C-1), 40.65 (C-8), 41.28 (CH2Cl), 42.40 (C-14), 46.96 (C-19), 49.41 (C-18), 50.43 (C-9), 55.37 (C-5), 56.53 (C-17), 65.74 (CH2Ph), 83.35 (C-3), 109.68 (C-29), 128.08, 128.26, 128.50 (Ph), 136.50 (Ph: C-1), 150.55 (C-20), 167.17 (C=O), 175.83 (C-28). Mass spectrum (ESI), m/z 529 [M + H – ClCH2CO2H]+ (calcd for C39H55ClO4, 622).
Preparation of 6a and 6b. A suspension of acid 3 (4.40 mmol) and K2CO3 (4.40 mmol) in DMF (18 mL) was treated dropwise with 1,2-dibromoethane or 1,3-dibromopropane (4.40 mmol) in DMF (2 mL) and stirred for 4 h. The K2CO3 was filtered off. The filtrate was evaporated. The residue was filtered through a layer of Al2O3. The filtrate was evaporated.
2-Bromoethyl Ester of 3-Oxolup-20(29)-en-28-oic Acid (6a). Yield 1.9 g (77%), amorph., \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +4° (c 0.25, CHCl3). IR spectrum (ν, cm–1): 1650, 1726. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.92 (3H, s, CH3-25), 0.97 (3H, s, CH3-26), 0.98 (3H, s, CH3-27), 1.02 (3H, s, CH3-24), 1.07 (3H, s, CH3-23), 1.69 (5H, s, CH3-30, m, H-15, H-18), 1.91 (3H, m, H-1, 21, 22), 2.24 (1H, td, J = 12.0, 4.8, H-13), 2.28 (1H, m, H-16), 2.44 (H, m, H-2), 3.02 (1H, td, J = 11.5, 5.3, H-19), 3.54 (2H, t, J = 6.0, CH2Br), 4.40, 4.42 (each 1H, dt, J = 12.0, 6.0, C(O)OCH 2CH2), 4.61, 4.74 (each 1H, s, H-29). 13C NMR spectrum (Table 1). Mass spectrum (APCI), m/z 561 [M + H]+ (calcd for C32H49BrO3, 560).
3-Bromopropyl Ester of 3-Oxolup-20(29)-en-28-oic Acid (6b). Yield 1.9 g (76%), mp 80–83°C, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –6° (c 0.40, CHCl3). IR spectrum (ν, cm–1): 1630, 1730. 1H NMR spectrum (300 MHz, CDCl3, δ, ppm, J/Hz): 0.92 (3H, s, CH3-25), 0.96 (3H, s, CH3-26), 0.97 (3H, s, CH3-27), 1.01 (3H, s, CH3-24), 1.06 (3H, s, CH3-23), 1.68 (5H, s, CH3-30, m, Hb-15, H-18), 1.88 (3H, m, H-1, 21, 22), 2.20 (2H, m, H-13, 16), 2.43 (2H, m, H-2), 3.00 (1H, m, H-19), 3.48 (2H, t, J = 6.5, CH2Br), 4.21 (2H, m, C(O)OCH2CH2), 4.64, 4.73 (each 1H, s, H-29) (only characteristic resonances are given). Table 1 lists the 13C NMR spectrum. Mass spectrum (APCI), m/z 575 [M + H]+ (calcd for C33H51BrO3, 574).
Tetrabutylammonium 2-{2-[3 β -Hydroxylup-20(29)-en-28-yloxy]-2-oxoethylamino}ethanesulfonate (7). A solution of NH2CH2CH2SO3 –Bu4N+ (0.16 g, 0.44 mmol) in DMF (4 mL) was treated with K2CO3 (0.06 g, 0.44 mmol), stirred for 10 min, treated dropwise with betulin chloroacetate (4, 0.23 g, 0.44 mmol) in DMF (5 mL), and stirred at 95°C for 3 h. The K2CO3 was filtered off. The filtrate was evaporated. The residue was chromatographed over SiO2 (C6H6–MTBE, 4:1; CHCl3–MeOH, 10:1) to afford 7 (0.25 g, 68%), amorph. IR spectrum (ν, cm–1): 1035, 1192, 1640, 1740, 3378. 1H NMR spectrum (500 MHz, CDCl3, δ, ppm, J/Hz): 0.67 (1H, d, J = 9.1, H-5), 0.76 (3H, s, CH3-24), 0.82 (3H, s, CH3-25), 0.96 (6H, s, CH3-23, 27), 1.01 (12H, t, J = 7.3, butyl: CH2CH2CH2CH 3), 1.03 (3H, s, CH3-26), 1.45 (10H, m, butyl: CH2CH2CH 2CH3, 2H-2), 1.67 (8H, m, butyl: CH2CH 2CH2CH3), 1.69 (3H, s, CH3-30), 1.76 (1H, dd, J = 12.2, 8.8, H-22), 1.83 (1H, d, J = 12.4, H-16), 1.95 (1H, m, H-21), 2.42 (1H, td, J = 10.5, 5.5, H-19), 3.03 (2H, t, J = 6.3, NCH2CH 2SO3), 3.13 (2H, t, J = 6.3, NCH 2CH2SO3), 3.18 (1H, dd, J = 11.2, 4.5, H-3), 3.32 (8H, t, J = 6.5, butyl: CH 2CH2CH2CH3), 3.48 (2H, s, OC(O)CH2), 3.88, 4.29 (each 1H, d, J = 11.0, H-28), 4.58, 4.68 (1H, s, H-29). Table 1 lists the 13C NMR spectrum. Mass spectrum (ESI), m/z 606 [M – N+Bu4]– (calcd for C50H92N2O6S, 848).
Tetrabutylammonium 2-{2-[17 β -Methoxycarbonyllup-20(29)-en-3 β -yloxy]-2-oxoethylamino}ethanesulfonate (8) was prepared analogously to 7. Yield 0.30 g (82%), mp 78–80°C, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –8.5° (c 0.43, CHCl3). IR spectrum (ν, cm–1): 1034, 1166, 1190, 1200, 1640, 1729, 3420. 1H NMR spectrum (500 MHz, CDCl3, δ, ppm, J/Hz): 0.76 (1H, d, J = 9.5, H-5), 0.82 (3H, s, CH3-25), 0.83 (3H, s, CH3-26), 0.91 (3H, s, CH3-24), 0.82 (3H, s, CH3-23), 0.95 (3H, s, CH3-27), 0.96 (1H, m, H-1), 1.01 (12H, t, J = 7.3, butyl: CH2CH2CH2CH 3), 1.14 (1H, m, H-15), 1.28 (1H, m, H-11), 1.36 (4H, m, 1H-6, 2H-7, 1H-21), 1.45 (4H, m, 1H-11, 15, 16, 22), 1.46 (2H, m, H-2), 1.47 (8H, sextet, J = 7.3, butyl: CH2CH2CH 2CH3), 1.49 (1H, m, H-6), 1.58 (1H, t, J = 11.3, H-18), 1.65 (9H, m, butyl: CH2CH 2CH2CH3, H-1), 1.65 (8H, m, butyl: CH2CH 2CH2CH3), 1.68 (3H, s, CH3-30), 1.88 (2H, m, H-21, 22), 2.21 (1H, td, J = 12.0, 5.5, H-13), 2.23 (1H, dd, J = 10.0, 3.3, H-16), 2.90 (1H, br.s, NH), 3.00 (1H, m, H-19), 3.01 (2H, t, J = 6.2, NCH2CH 2SO3), 3.12 (2H, t, J = 6.2, NCH 2CH2SO3), 3.31 (8H, t, J = 6.5, butyl: CH 2CH2CH2CH3), 3.45 (2H, s, OC(O)CH2), 3.67 (3H, s, C(O)OCH3), 4.50 (1H, dd, J = 11.0, 5.5, H-3), 4.60, 4.73 (each 1H, s, H-29). Table 1 lists the 13C NMR spectrum. Mass spectrum (ESI), m/z 634 [M – N+Bu4]– (calcd for C51H92N2O7S, 876).
Preparation of 9, 10a, and 10b. General Method. A suspension of N-methyltaurine (0.19 mmol) in DMF (3 mL) was treated with K2CO3 (0.19 mmol), stirred for 15 min, treated dropwise with chloroacetate 4, 5a, or 5b (0.19 mmol) in DMF (2 mL), and stirred at 95°C for 50 h (for 9) or 6 h (for 10a and 10b). The K2CO3 was filtered off. The filtrate was evaporated to dryness. The resulting oily residue was triturated with hexane. The resulting solid was filtered off. Solid compound 9 was rinsed with MTBE and dried in vacuo (KOH, 80°C). Compounds 10a and 10b that were obtained after hexane work-up were chromatographed over SiO2 (C6H6–MTBE, 4:1; CHCl3–MeOH, 4:1).
2-({2-[3 β -Hydroxylup-20(29)-en-28-yloxy]-2-oxoethyl}(methyl)amino)ethanesulfonic Acid (9). Yield 0.11 g (90%), amorph., \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +9.5° (c 0.64, CH3OH). IR spectrum (ν, cm–1): 1038, 1192, 1213, 1637, 1653, 1748, 3420. 1H NMR spectrum (500 MHz, DMSO-d6, δ, ppm, J/Hz): 0.62 (1H, d, J = 9.8, H-5), 0.65 (3H, s, CH3-24), 0.76 (3H, s, CH3-25), 0.83 (1H, m, Ha-1), 0.87 (3H, s, CH3-23), 0.94 (3H, s, CH3-27), 0.99 (5H, s, CH3-26, m, Ha-12, 15), 1.09 (1H, m, Ha-22), 1.20 (1H, m, Ha-11), 1.26 (2H, m, H-9, Ha-16), 1.34 (5H, m, Ha-6, 21, 2H-7, Hb-11), 1.44 (3H, m, 2H-2, Hb-6), 1.54 (1H, m, Hb-1), 1.57 (1H, m, H-18), 1.60 (2H, m, H-13, Hb-12), 1.65 (4H, s, CH3-30, m, Hb-15), 1.72 (1H, m, Hb-22), 1.74 (1H, m, Hb-16), 1.91 (1H, m, Hb-21), 2.48 (1H, m, H-19), 2.66 (3H, s, NCH3), 2.84 (2H, t, J = 7.5, NCH2CH 2SO3), 2.96 (1H, dd, J = 10.0, 5.3, H-3), 3.21 (2H, m, NCH 2CH2SO3), 3.28 (1H, d, J = 10.8, Ha-28), 3.98 (2H, s, OC(O)CH2), 4.37 (1H, d, J = 10.8, Hb-28), 4.57, 4.71 (each 1H, s, H-29), 8.73 (1H, br.s, SO3H). Table 1 lists the 13C NMR spectrum. Mass spectrum (ESI), m/z 620 [M – H]–(calcd for C35H59NO6S, 621).
2-({2-[17 β -Methoxycarbonyllup-20(29)-en-3 β -yloxy]-2-oxoethyl}(methyl)amino)ethanesulfonic Acid (10a). Yield 0.08 g (66%), amorph., \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) –8° (c 0.65, CH3OH). IR spectrum (ν, cm–1): 1045, 1174, 1188, 1201, 1640, 1729, 3446. 1H NMR spectrum (300 MHz, DMSO-d6, δ, ppm, J/Hz): 0.79 (10H, m, H-5, s, CH3-23, 24, 25), 0.83 (3H, s, CH3-26), 0.94 (3H, s, CH3-27), 1.64 (3H, s, CH3-30), 2.12 (1H, m, H-16), 2.24 (3H, s, NCH3), 2.63 (2H, m, NCH2CH 2SO3), 2.75 (2H, m, NCH 2CH2SO3), 2.90 (1H, m, H-19), 3.23 (2H, s, OC(O)CH2), 3.58 (3H, s, C(O)OCH3), 4.40 (1H, dd, J = 10.5, 5.3, H-3), 4.56, 4.69 (each 1H, s, H-29). Table 1 lists the 13C NMR spectrum. Mass spectrum (ESI), m/z 648 [M – H]– (calcd for C36H59NO7S, 649).
2-({2-[17 β -Benzyloxycarbonyllup-20(29)-en-3 β -yloxy]-2-oxoethyl}(methyl)amino)ethanesulfonic Acid (10b). Yield 0.09 g (64%), amorph., \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +11° (c 0.38, CH3OH). IR spectrum (ν, cm–1): 1041, 1129, 1149, 1191, 1231, 1640, 1719, 1724, 3300. 1H NMR spectrum (500 MHz, DMSO-d6, δ, ppm, J/Hz): 0.69 (3H, s, CH3-24), 0.78 (3H, s, CH3-25), 0.79 (7H, m, H-5, s, CH3-23, 26), 0.92 (4H, m, Ha-1, s, CH3-27), 0.97 (1H, m, Ha-12), 1.03 (1H, m, Ha-15), 1.12 (1H, m, Ha-11), 1.22 (2H, m, Ha-7, Hb-15), 1.27 (2H, m, Ha-6, H-9), 1.32 (3H, m, Hb-7, 11, Ha-21), 1.42 (2H, m, Hb-6, Ha-16), 1.47 (1H, m, Ha-22), 1.55 (1H, m, Ha-2), 1.57 (2H, m, Hb-2, H-18), 1.60 (2H, m, Hb-1, 12), 1.64 (3H, s, CH3-30), 1.75 (1H, m, Hb-21), 1.80 (1H, m, Hb-22), 2.11 (1H, td, J = 11.0, 4.0, H-13), 2.17 (1H, d, J = 13.0, Hb-16), 2.26 (3H, s, NCH3), 2.62 (2H, m, NCH2CH 2SO3), 2.78 (2H, m, NCH 2CH2SO3), 2.94 (1H, td, J = 11.0, 5.5, H-19), 3.22, 3.26 (each 1H, d, J = 17.3, OC(O)CH2), 4.41 (1H, dd, J = 10.5, 5.3, H-3), 4.56, 4.68 (each 1H, s, H-29), 5.08, 5.12 (each 1H, d, J = 12.3, CH 2Ph), 7.35 (5H, s, Ph). Table 1 lists the 13C NMR spectrum. Mass spectrum (ESI), m/z 724 [M – H]– (calcd for C42H63NO7S 725).
Preparation of 11a and 11b. A suspension of the Na-salt of N-methyltaurine (0.14 g, 0.89 mmol) and K2CO3 (0.12 g, 0.89 mmol) in DMF (8 mL) was stirred under Ar for 15 min, treated dropwise with the appropriate bromoalkyl ester 6a or 6b (0.50 g, 0.89 mmol) in DMF (12 mL), and stirred for 50 h at 95°C. The K2CO3 was filtered off. The filtrate was evaporated. The oil residue was triturated with hexane. The solution was decanted. The solid was rinsed with MTBE (TLC monitoring) and dried (KOH, 80°C).
Sodium 2-({2-[3-Oxolup-20(29)-en-17 β -carbonyloxy]ethyl}(methyl)amino)ethanesulfonate (11a). Yield 0.42 g (74%), mp 170–172°C, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +15.3° (c 0.45, CH3OH). IR spectrum (ν, cm–1): 1200, 1720, 3420. 1H NMR spectrum (500 MHz, CD3OD, δ, ppm, J/Hz): 0.93 (3H, s, CH3-25), 0.98 (3H, s, CH3-26), 1.01 (6H, s, CH3-24, 27), 1.05 (3H, s, CH3-23), 1.10 (1H, qd, J = 12.5, 5.0, H-12), 1.20 (1H, m, Ha-15), 1.30 (1H, dt, J = 12.5, 5.0, Ha-11), 1.40 (1H, m, H-5), 1.46 (10H, m, 1H-1, 6, 16, 21, 22, 2H-7, H-9, 1H-11, 15), 1.50 (1H, m, H-6), 1.66 (1H, t, J = 12.5, H-18), 1.68 (3H, s, CH3-30), 1.73 (1H, m, H-12), 1.91 (2H, m, H-1, 22), 2.28 (2H, m, H-16, 21), 2.30 (1H, m, H-13), 2.33 (3H, s, NCH3), 2.48 (2H, m, H-2), 2.70 (2H, m, C(O)OCH2CH 2)), 2.93 (2H, m, NCH 2CH2SO3), 3.00 (1H, m, H-19), 3.03 (2H, m, NCH2CH 2SO3), 4.18, 4.24 (each 1H, dt, J = 11.5, 5.3, C(O)OCH 2CH2), 4.59, 4.71 (each 1H, s, H-29). Table 1 lists the 13C NMR spectrum. Mass spectrum (ESI), m/z 618 [M – Na]– (calcd for C35H56NNaO6S, 641).
Sodium 2-({2-[3-Oxolup-20(29)-en-17 β -carbonyloxy]propyl}(methyl)amino)ethanesulfonate (11b). Yield 0.40 g (69%), mp 162–164°C, \( {\left[\upalpha \right]}_{\mathrm{D}}^{20} \) +15.5° (c 0.54, CH3OH). 1H NMR spectrum (300 MHz, CD3OD, δ, ppm, J/Hz): 0.95 (3H, s, CH3-25), 0.99 (3H, s, CH3-26), 1.02 (3H, s, CH3-24), 1.03 (3H, s, CH3-27), 1.06 (3H, s, CH3-23), 1.70 (5H, s, CH3-30, m, 1H-12, H-18), 1.88 (4H, m, 1H-1, 22, C(O)OCH2CH 2CH2), 2.29 (3H, m, H-13, 1H-16, 21), 2.38 (3H, s, NCH3), 2.51 (4H, m, 2H-2, C(O)OCH2CH2CH 2), 2.92 (2H, m, NCH 2CH2SO3), 3.00 (3H, m, NCH2CH 2SO3, H-19), 4.12 (2H, m, C(O)OCH 2CH2CH2), 4.61, 4.73 (each 1H, s, H-29) (only characteristic resonances are given). Table 1 lists the 13C NMR spectrum. Mass spectrum (ESI), m/z 632 [M – Na]– (calcd for C36H58NNaO6S, 655).
Screening for Toxicity in vitro . The toxicity of the compounds was studied using a continuous culture of Madin-Darby canine kidney cells (MDCK) that was obtained initially from the Centers for Disease Control and Prevention (CDC, Atlanta, USA). Toxicity in vitro was assessed using the MTT assay, i.e., reduction of the dye thiazolyl blue tetrazolium bromide (MTT) by cells in culture. The experiments with the cell culture were conducted as follows. A weighed portion (5 mg) of compound in a sterile tube was diluted with DMSO to a concentration of 1 mg/mL (stock solution). Then, six sequential two-fold dilutions (100, 50, 25, 12.5, 6, and 3 μg/mL) with MDCK growth medium (Biolot, St. Petersburg) produced the solutions used to determine the toxicity. The test was performed in triplicate for each concentration. A 1-d culture of MDCK cells grown in 96-well plates was inspected visually using an inverted microscope for the integrity of the monolayer. The plates were rinsed twice with medium without serum. The test compound (100 μL per well) of the appropriate concentration was placed into each well. The plates were incubated for 72 h at 37°C with 5% CO2. Test results were recorded visually by evaluating the integrity of the cell monolayer as compared with a control.
The MTT assay used 96-well plates and the standard protocol [25]. Optical density was recorded at 550 nm on a Varioscan microplate reader (Thermo Fisher). The median cytotoxic dose (CTD50) was determined by linear regression of the photometric data using the Excel 2010 program. The regression equations were valid because R2 > 0.9.
References
P. A. Krasutsky, Nat. Prod. Rep., 23, 919 (2006).
R. Csuk, Expert Opin. Ther. Pat., 24 (8), 913 (2014).
P. Yogeeswari and D. Sriram, Curr. Med. Chem., 12 (6), 657 (2005).
H. Wang, R. Xu, Y. Shi, L. Si, P. Jiao, Z. Fan, X. Han, X. Wu, X. Zhou, F. Yu, Y. Zhang, D. Zhou, and S. Xiao, Eur. J. Med. Chem., 110, 376 (2016).
H. Ripps and W. Shen, Mol. Vision, 18, 2673 (2012).
N. Chen and J. Xu, Tetrahedron, 68 (11), 2513 (2012).
R. C. Gupta, T. Win, and S. Bittner, Curr. Med. Chem., 12 (17), 2021 (2005).
A. A. Shtro, A. V. Slita, L. A. Karpinskaya, A. V. Galochkina, and V. V. Zarubaev, Lechashchii Vrach, 10, 52 (2012).
S. Q. Yang, M. Froeyen, E. Lescrinier, P. Marliere, and P. Herdewijn, Org. Biomol. Chem., 9 (1), 111 (2011).
M. G. Mutchnick, M. N. Ehrinpreis, J. L. Kinzie, and R. R. Peleman, Antiviral Res., 24 (2–3), 245 (1994).
J. Yoon, A. Jekle, R. Najafi, F. Ruado, M. Zuck, B. Khosrovi, B. Memarzadeh, D. Debabov, L. Wang, and M. Anderson, Antiviral Res., 92 (3), 470 (2011).
T. P. Shiau, E. Low, B. Kim, E. D. Turtle, C. Francavilla, D. J. R. O’Mahony, L. Friedman, L. D’Lima, A. Jekle, D. Debabov, M. Zuck, N. J. Alvarez, M. Anderson, R. Najafi, and R. K. Jain, Bioorg. Med. Chem. Lett., 23 (20), 5650 (2013).
F. Kong, N. Zhao, M. M. Siegel, K. Janota, J. S. Ashcroft, F. E. Koehn, D. B. Borders, and G. T. Carter, J. Am. Chem. Soc., 120 (51), 13301 (1998).
Y. Takahashi, J.-R. Nakijiquinones, M. Ushio, T. Kubota, S. Yamamoto, J. Fromont, and J. Kobayashi, J. Nat. Prod., 73 (3), 467 (2010).
A. Aiello, E. Fattorusso, P. Luciano, A. Macho, M. Menna, and E. Munoz, J. Med. Chem., 48 (9), 3410 (2005).
A. Barthel, S. Stark, and R. Csuk, Tetrahedron, 64 (39), 9225 (2008).
D. S. H. L. Kim, Z. Chen, T. Nguyen, J. M. Pezzuto, S. Qiu, and Z.-Z. Lu, Synth. Commun., 27 (9), 1607 (1997).
N. V. Uzenkova, N. I. Petrenko, M. M. Shakirova, E. E. Shul’ts, and G. A. Tolstikov, Chem. Nat. Compd., 41, 692 (2005).
X. Y. Wang, S. Y. Zhang, J. Li, H. N. Liu, X. Xie, and F. J. Nan, Acta Pharm. Sin., 35, 1463 (2014).
J. W. Schick and E. F. Degering, Ind. Eng. Chem., 39 (7), 906 (1947).
D. G. I. Kingston and Z.-Y. Zhao, EP Pat. 0473326 A1, Mar. 4, 1992.
X. Fang, J. Ling, F. Liu, and Y. Chen, CN Pat. 102675160A, Sept. 19, 2012.
M. S. Yunusov, N. G. Komissarova, and N. G. Belenkova, RU Pat. No. 2,270,201, Feb. 20, 2006; Byull. Izobret., No. 5, 1 (2006).
H. Kommera, G. N. Kaluderovic, J. Kalbitz, and R. Paschke, Arch. Pharm. Chem. Life Sci., 343 (8), 449 (2010).
E. Borenfreund, H. Babich, and N. Martin-Alguacil, Toxicol. in Vitro, 2 (1), 1 (1988).
Acknowledgment
The work was supported financially by a grant from the Russian Science Foundation (Project No. 14-13-01307).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2017, pp. 772–778.
Rights and permissions
About this article

Cite this article
Komissarova, N.G., Dubovitskii, S.N., Shitikova, O.V. et al. Synthesis of Conjugates of Lupane-Type Pentacyclic Triterpenoids with 2-Aminoethane- and N-Methyl-2-Aminoethanesulfonic Acids. Assessment of in vitro Toxicity. Chem Nat Compd 53, 907–914 (2017). https://doi.org/10.1007/s10600-017-2153-6
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-017-2153-6