
A new pyranoxanthone, buxixanthone (1), was isolated from the stem bark of Calophyllum buxifolium along with three other chemical constituents, ananixanthone (2), pyranojacareubin (3), and macluraxanthone (4). The structures of these compounds were confirmed through spectroscopic analysis, which included 1D and 2D NMR, GC/MS, and IR experiments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The Guttiferae family is a well-known group of trees and can be easily found in tropical Asia and Africa [1]. It consists of four genera, one of which is Calophyllum. The Malay local name for Calophyllum is “bitangor” [2, 3]. Previous work has shown the secondary metabolites from this genus to possess various bioactivities such as anti-HIV-1 [4], antimicrobial [5], and cytotoxic activities [6]. Plants from this genus are rich in xanthones, coumarin, flavanoid, and terpenoids. Some of these compounds have shown good biological activities such as antifungal, antimalarial, and antioxidant. In view of the many pharmaceutical potentials of this genus, we focus our recent work on the stem bark of Calophyllum buxifolium. This has led to the isolation of one new xanthone, buxixanthone (1), along with three other chemical constituents, ananixanthone (2), pyranojacareubin (3), and macluraxanthone (4). There are no previous reports on the chemistry of Calophyllum buxifolium.
Compound 1 was obtained as yellow crystals with a melting point of 180–183°C and reacted positively with FeCl3 reagent, indicating the presence of a phenolic group. The EI-MS analysis showed a molecular mass of 392 for compound 1, which corresponds to the molecular formula C24H24O5. The FTIR spectrum showed absorptions of free hydroxyl at 3410 cm–1, conjugated carbonyl at 1710 cm–1, and aromatic ring at 1590 cm–1. Meanwhile, the UV spectrum confirmed compound 1 to have a xanthone skeleton with UV adsorptions at 380, 286, 235, and 207 nm.

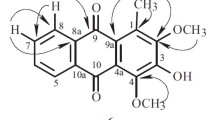
The 1H NMR experiment showed two singlet signals at δ 13.67 and 3.86, indicating chelated hydroxyl and methoxyl moieties in compound 1. The HMBC correlations between 1-OH and δ 158.18 (C-1) and 104.28 (C-9a) in the HMBC spectrum suggested the attachment of the hydroxyl group at C-1. On the other hand, the position of the methoxyl at C-6 was confirmed by the methoxyl protons giving a correlation with δ 153.59 (C-6) in the HMBC spectrum (Fig. 1). One triplet signal at δ 5.22 (1H, J = 6.8 Hz, H-17), a doublet signal at δ 4.15 (2H, J = 6.9 Hz, H-16), and two singlets at 1.83 (3H, H-19) and 1.67 (3H, H-20) were assigned to the isoprene moiety in this compound. The COSY experiment for compound 1 gave couplings between H-16 and H-17. The position for this prenyl group was suggested to be at C-8 as there are correlations between H-16 and δ 131.96 (C-8, s), and 118.73 (C-7, d) in the HMBC spectrum.

HMBC correlations of buxixanthone (1).
Meanwhile, resonances typical of a 2,2-dimethylpyrano system with a 6H-singlet at δ 1.46 and two AB doublets (J = 9.16 Hz) located at δ 6.73 and 5.74 were observed in the spectrum of compound 1. These two doublets were assigned to the protons attached to C-11 and C-12, respectively. These two signals were also seen to be coupled to each other in the COSY experiment. The long-range 3J correlations of H-12 (δ 5.74) with C-4 (δ 104.28) in the HMBC spectrum indicated that the pyrano ring was fused to the xanthone ring B at C-4 and to C-3 through oxygen. This was confirmed by the observation of a 3J long range correlation between H-11 and C-3. The very downfield carbon signal at δ 183.40 (C-9) in the 13C spectrum was assigned to the conjugated carbonyl carbon in this compound. The remaining two proton signals, which indicated a meta coupling of J = 2.3 Hz at δ 7.25 (H-5) and 7.26 (H-7) in the 1H NMR spectrum, were assigned to the protons at C-5 and C-7. Both H-5 and H-7 gave 2J HMBC correlations with C-6. All the carbons in the compound were assigned to their respective protons by their 1J correlations in the HMQC spectrum. This compound was confirmed to consist of six methines, one methylene, four methyls, one methoxyl, and 12 quartenary carbons from the DEPT analysis. Hence the compound was elucidated to be 1-hydroxy-6-methoxy-8-(3-methylbut-2-enyl)-6′6′-dimethylpyrano[2′,3′:3,4]xanthone and given the trivial name buxixanthone (1).
Experimental
General. 1H NMR spectra were recorded on JEOL JNM-ECX500 (500 MHz) or JEOL ECA400 (400 MHz) spectrometers. 13C and DEPT NMR spectra were obtained in both instruments operating at 125 and 100 MHz, respectively. Tetramethylsilane (TMS) was used as an internal standard, and deuterated solvents were used in the analysis. Infrared spectra were obtained through the universal attenuated total reflection (UATR) technique on a PerkinElmer 100 Series FT-IR spectrometer. Mass spectroscopic data were recorded using a Shidmadzu GC-MS model QP2010 Plus spectrometer. Melting points were obtained on a Leica Galen III instrument. Ultraviolet spectra were recorded in ethanol (EtOH) on a Shimadzu UV-160A UV-Visible recording spectrophotometer.
Plant Material. The stem bark samples of Calophyllum buxifolium voucher specimens (Voucher specimen No. RG 5020) were deposited in the Herbarium of the Biology Department, Faculty of Science, Universiti Putra Malaysia.
Extraction and Isolation. The dried and powdered stem bark of Calophyllum buxifolium (1.8 kg) was extracted with n-hexane and ethyl acetate by the conventional soaking method for 48 h at room temperature. The extracts were then evaporated to dryness under vacuum to give 15.7 g of n-hexane extract and 28.5 g of ethyl acetate extract.
Approximately 10 g of n-hexane extract was subjected to column chromatography using hexane–ethyl acetate (3:7) and chloroform–methanol (9:1) as eluting system to give 15 fractions. Fraction 3 was then repeatedly purified by column chromatography and gave yellow crystals of ananixanthone (2) (3 mg). Meanwhile, further purification of fraction 7 resulted in macluraxanthone (4) (3 mg) as a pale yellow crystals. Fractions 8 to 11 were pooled together and rechromatographed in another column of silica gel and eluted with petroleum ether–ethyl acetate (6:4) to give seven more fractions. Fraction 4 was washed with hexane to give yellow needle crystals of pyranojacareubin (3) (2 mg).
Twenty grams of the ethyl acetate extract was chromatographed using column chromatography with petroleum ether–ethyl acetate (5:5) and chloroform–acetone (4:6) as the mobile phase to give 20 fractions. Fractions 5 to 8 were pooled together and resubjected to gravity column chromatography using silica gel and eluted with hexane–acetone (1:1) to give another five fractions. Fractions 3 and 4 were then further purified using Sephadex lipophilic LH-20 with methanol (10:0) and methanol–chloroform (9.5:0.5) as eluting solvents to afford a new xanthone, buxixanthone (1) (4 mg).
Buxixanthone (1). Yellow crystals, mp 180–183°C. UV (EtOH, λmax, nm): 380, 286, 235, 207. IR (νmax, cm–1): 3420, 1710, 1590. EI-MS (m/z, I rel., %): 392 (39), 378 (16), 377 (61), 359 (14), 349 (100), 307 (11). 1H NMR (500 MHz, CDCl3, δ, ppm, J/Hz): 13.67 (1H, s, 1-OH), 7.26 (1H, d, J = 2.3, H-7), 7.25 (1H, d, J = 2.3, H-5), 6.73 (1H, d, J = 9.16, H-11), 6.24 (1H, s, H-2), 5.74 (1H, d, J = 9.16, H-12), 5.22 (1H, t, J = 6.9, H-17), 4.15 (2H, d, J = 6.9, H-16), 3.86 (3H, s, OCH3), 1.83 (3H, s, H-19), 1.67 (3H, s, H-20), 1.46 (6H, s, H-14, 15). 13C NMR (125 MHz, CDCl3, δ, ppm): 183.40 (C-9), 160.41 (C-3), 158.18 (C-1), 155.66 (C-4a), 153.59 (C-6), 151.73 (C-10a), 132.14 (C-18), 131.96 (C-8), 127.16 (C-12), 122.82 (C-17), 118.73 (C-7), 115.98 (C-5), 115.92 (C-8a), 115.79 (C-11), 104.28 (C-4), 104.28 (C-9a), 94.13 (C-2), 78.11 (C-13), 56.84 (6-OCH3), 28.42 (C-14), 28.42 (C-15), 26.06 (C-20), 25.66 (C-16), 18.17 (C-19).
Ananixanthone (2). Yellow needle crystals, mp 168–170°C [Lit. 170–171°C] [7]. UV (EtOH, λmax, nm): 337, 302, 289, 261. IR (νmax, cm–1): 3203, 1646, 1575. EI-MS m/z: 378, 363, 364, 335, 154. 1H NMR (500 MHz, CDCl3, δ, ppm, J/Hz): 13.19 (1H, s, 1-OH), 7.77 (1H, dd, J = 8.02, 1.5, H-8), 7.30 (1H, dd, J = 8.02, 1.5, H-6), 7.25 (1H, t, J = 8.02, H-7), 6.79 (1H, d, J = 10.1, H-11), 5.70 (1H, d, J = 10.1, H-12), 5.24 (1H, t, J = 6.9, H-17), 3.36 (2H, d, J = 6.9, H-16), 1.81 (3H, s, H-20), 1.68 (3H, s, H-19), 1.48 (6H, s, H-14, 15).
Pyranojacareubin (3). Yellow crystals, mp 261–263°C [Lit. 259–261°C] [8]. UV (EtOH, λmax, nm) (log ε): 244.5 (0.31), 261.0 (0.33), 317.5 (0.19). IR (νmax, cm–1): 3454, 2968, 1640, 1610. EI-MS m/z: 392, 378, 377, 181, 41. 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 13.38 (1H, s, 1-OH), 7.62 (1H, s, H-8), 6.67 (1H, d, J = 10.1, H-12), 6.42 (1H, d, J = 10, H-17), 6.40 (1H, s, H-4), 5.76 (1H, d, J = 10, H-16), 5.61 (1H, d, J = 10.1, H-11), 1.52 (6H, s, H-19, 20), 1.47 (6H, s, H-14, 15).
Macluraxanthone (4). Pale yellow crystals, mp 175–178°C [Lit. 170–172°C] [9]. UV (EtOH, λmax, nm): 335, 270, 245. IR (νmax, cm–1): 3309, 2926, 1643, 1583. EI-MS m/z: 394, 379, 365, 339, 321, 162. 1H NMR (400 MHz, acetone-d6, δ, ppm, J/Hz): 13.87 (1H, s, 1-OH), 7.58 (1H, d, J = 10, H-8), 6.98 (1H, d, J = 10, H-7), 6.68 (1H, d, J = 10.1, H-11), 6.52 (1H, dd, J =18, 11, H-19), 5.68 (1H, d, J = 10.1, H-12), 5.05 (1H, dd, J = 18, 1, H-20Z), 4.87 (1H, dd, J = 11, 1, H-20E), 1.72 (6H, s, H-17, 18), 1.46 (6H, s, H-14, 15).
References
N. M. Nasir, M. Rahmani, K. Shaari, N. K. Kassim, J. Go R. Stanslas, and J. J. Ethel, Sains Malaysiana, 42 (9), 1261 (2013).
E. J. H. Corner, Wayside Trees of Malaya, The Malayan Nature Society, Kuala Lumpur, Malaysia, 1988.
T. C. Whitmore, Tree Flora of Malaya: A Manual for Foresters, Vol. II, Longman, London, 1973, pp. 162–195.
A. D. Patil, A. J. Freyer, D. S. Eggleston, R. C. Haltiwanger Bean, P. B. Taylor, M. J. Caranfa, A. L. Breen, H. R. Bartus, R. K. Johnson, R. P. Hertzberg, and J. W. Westley, J. Med. Chem., 36, 4131 (1993).
M. C. Yimdjo, A. G. Azebaze, A. E. Nkengfack, A. M Meyer, B. Bodo, and Z. T. Fomum, Phytochemistry, 65 (20), 2789 (2004).
Q. Xiao, Y. B. Zeng, W. L. Mei, Y. X. Zhao, Y. Y. Deng, and H. F. Dai, J. Asian Nat. Prod., 10, 993 (2008).
C. B. Joaquim, S. P. A. Mara, and S. N. Marcelino, Phytochemistry, 49 (4), 1159 (1998).
L. J. Harrison, L. S. Leong, G. L. Sia, K. Y. Sim, and H. T. W. Tan, Phytochemistry, 33 (3), 727 (1993).
M. Iinuma, H. Tosa, T. Tanaka, and S. Yonemori, Phytochemistry, 35 (2), 527 (1994b).
Acknowledgment
The authors wish to express their gratitude for financial support from UPM and MOSTI under the Agri Science Fund. The SBC is also acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Prirodnykh Soedinenii, No. 5, September–October, 2016, pp. 693–695.
Rights and permissions
About this article

Cite this article
bin Daud, S., Ee, G.C.L., Malek, E.A. et al. A New Pyranoxanthone from the Stem Bark of Calophyllum buxifolium . Chem Nat Compd 52, 807–809 (2016). https://doi.org/10.1007/s10600-016-1783-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10600-016-1783-4