

A simple one-step protocol for the synthesis of 3-amino-1-benzothiophene-2-carbonitriles from commercially or synthetically readily available S-(cyanomethyl) O-ethyl carbodithionate and 2-fluorobenzonitriles was developed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
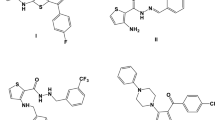
The 1-benzothiophene scaffold forms the basic structural fragment of a number of antimicrobial,1,2 antitumor,3 antifungal4 drugs, hormonal modulators,5 and antioxidants6 (Fig. 1). An analysis of literature data7 revealed that the biological activity of benzothiophenes is achieved primarily via functionalization at positions 2 and 3 of the thiophene ring. In this regard, novel approaches to the preparation of 2,3-substituted 1-benzothiophenes that combine functional groups of different nature are relevant.

Biologically active benzothiophene derivatives.
The synthesis of such compounds is realized mainly via a tandem of nucleophilic substitution – intramolecular condensation reactions. Sulfides,8 thiols,9 and thiourea are employed as S-nucleophiles.10 Recyclization of 1,2-benzothiazoles by the action of carbanions present a fundamentally different approach.11 However, the presented methods are limited either by the low availability of precursors or by the reaction conditions (microwave irradiation, low tolerance to substituents). In this work we proposed a simple one-step method for the preparation of 3-amino-2-cyano-1-benzothiophenes from benzonitriles and xanthates which are commercially or synthetically readily available and stable precursors.
We were prompted to study the reaction of xanthates with 2-fluorobenzonitriles by the synthesis of benzothiophene analogs, thieno[2,3-b]pyridines, from 2-chloro-3-cyanopyridine reported by us earlier.12 The involvement of more accessible but less electron-deficient 2-fluorobenzonitriles required more careful selection of the reaction conditions (Table 1), but made it possible to significantly expand the synthetic applicability of this approach.
By optimizing the reaction conditions using 2-fluorobenzonitrile (1a) as a model precursor, we found that the reaction did not proceed under the previously proposed conditions,12 that is, by the action of Cs2CO3 in MeOH. It was possible to obtain the target product in the polar aprotic solvent DMSO in the presence of DBU, a potent non-nucleophilic base. It was found that under these conditions, concurrently with the formation of the target product, a fairly rapid decomposition of the starting xanthate 2 was observed – it could not detected by TLC in the reaction mixture after 2 h. Attempts to increase the starting amount of xanthate 2 led to a slight increase in the yield, with some of the starting 2-fluorobenzonitrile (1a) remaining unreacted. In contrast, gradual addition of excess xanthate 2 to the reaction mixture allowed us to achieve complete conversion of the nitrile and obtain benzothiophene 3a in 81% yield. Thus, it turned out to be optimal to gradually add excess xanthate 2 (4 equiv by 1 equiv every 2 h) to the reaction mixture initially containing excess base (4.2 equiv of DBU).
Extension of this technique to 2-fluorobenzonitriles 1a–g made it possible to obtain benzothiophenes 3a–g in yields of up to 95% (Scheme 1). The exception was derivative 3g containing an electron donor substituent, the yield of which was only 30%. However, isolation of the unreacted precursor showed that in this case incomplete conversion was observed, and therefore a higher yield could be achieved by adding additional amounts of xanthate 2. The absence of side products indicated a high tolerance of the reaction conditions to the presence of ester and aniline functional groups, which is of great importance for the synthesis of biologically active compounds.

Scheme 1
The proposed reaction mechanism (Scheme 2) involves the formation of two anionic intermediates from xanthate 2. Carbanion A is formed directly from xanthate 2 by the action of a strong base. The action of carbanion A on another molecule of xanthate 2 leads to thiolate B, which, in addition to the main reaction, could participate in the side process of degradation of xanthate 2. This was indirectly confirmed by the detection of thioester 4 in the reaction mixture by 1H NMR spectroscopy. Two reaction routes are possible for intermediates A and B leading to the formation of the target product 3a.

Scheme 2
The first route for the formation of benzothiophene (path a) begins with a nucleophilic attack of carbanion A on the nitrile group of precursor 1a. Similar processes involving intermediate A on the example of carbonyl compounds can be found in the literature.13 The resulting intermediate A contains a carbodithionate group, which eliminates the thiolate ion by the action of a nucleophile. The role of the nucleophile could be played by intermediates A, B, or other anions present in the reaction mixture. Subsequent intramolecular nucleophilic substitution leads to product 3a.
The second route (path b) is realized via nucleophilic substitution involving intermediate A and a subsequent intramolecular Thorpe–Ziegler cyclization with the formation of the target benzothiophene 3a. Thus, the two possible reaction routes differ in the sequence of formation of the C–C and C–S bonds of the thiophene ring. The assumption that xanthate 2 is involved in several processes explains why a gradual addition of xanthate 2 provides a greater yield than its introduction in one portion. With a single addition of xanthate 2, most of it decomposes to product 4 or other side products and does not lead to the formation of benzothiophene 3a.
To conclude, we have developed a simple protocol for the preparation of 3-amino-1-benzothiophene-2-carbonitriles from readily available reagents. The proposed synthetic approach does not require special reaction conditions and demonstrates tolerance of a number of functional groups and therefore could be employed to obtain a variety of complexly substituted biologically active compounds.
Experimental
1H and 13C NMR spectra were registered on a Bruker Avance III 800 (800 and 201 MHz, respectively) and Bruker Fourier 300 (300 and 75 MHz, respectively) spectrometers in DMSO-d6 and CDCl3. TMS or the residual solvent signal (DMSO-d6: 2.50 ppm for 1H nuclei, 39.5 ppm for 13C nuclei; CDCl3: 7.26 ppm for 1H nuclei, 77.2 ppm for 13C nuclei) served as internal standards. High-resolution mass spectra were recorded on an AB Sciex TripleTOF 5600+ instrument with electrospray ionization. Melting points were determined on an SMP 30 apparatus.
Reagents supplied by Acros Organics were used without additional purification; freshly distilled solvents were used to carry out the reactions. S-(Cyanomethyl) O-ethyl carbodithionate (2) was obtained according to a literature method.14
S-(Cyanomethyl) O-ethyl carbodithionate (2). Potassium O-ethyl xanthate (6.50 g, 40.6 mmol) was dissolved in Me2CO (100 ml). The resulting solution was cooled to 0°C (in an ice bath), and chloroacetonitrile (2.55 g, 2.1 ml, 33.8 mol) was added dropwise with vigorous stirring over 3 min. The reaction mixture was stirred for 2 h at room temperature, then the solvent was evaporated on a rotary evaporator. The residue was dissolved in EtOAc (100 ml), the solution was treated with H2O (100 ml), then with saturated aqueous NaCl solution (3×50 ml), and finally dried over Na2SO4. The solvent was evaporated, the product was purified by flash chromatography (eluent hexane–EtOAc, 9:1). Yield 5.05 g (76%), clear pale-yellow oil. 1H NMR spectrum (800 MHz, CDCl3), δ, ppm (J, Hz): 4.72 (2H, q, J = 7.1, CH2); 3.89 (2H, s, CH2); 1.47 (3H, t, J = 7.1, CH3). The spectral data coincide with those published in the literature.15
Optimization of the synthesis of 3-amino-1-benzothiophene-2-carbonitrile (3a) (Table 1). 2-Fluorobenzonitrile 1a (10 mg, 83 μmol, 1 equiv) was dissolved in the corresponding solvent (1 ml), and the indicated amounts of xanthate 2 and base were added. The reaction mixture was stirred for the specified time. In entries 14 and 15 (Table 1), xanthate 2 was added to the reaction mixture in portions: 1 equiv three and four times, respectively, in 2-h intervals. The progress of the reactions was monitored by the disappearance of xanthate 2 in the reaction mixture by TLC (eluent hexane–EtOAc, 9:1, Rf 0.22). After the specified period of time, the reaction mixture was diluted with saturated aqueous NaCl solution (50 ml) and extracted with EtOAc (50 ml). The organic phase was dried over Na2SO4, the solvent was evaporated to dryness on a rotary evaporator, and the residue was analyzed by 1H NMR spectroscopy. The yield of the product was determined by assuming the sum of the integral intensities of 2-fluorobenzonitrile 1a and product 3a in the range of 7.89–8.15 ppm as 100%. In entries 8–15, thioester 4 was detected in the 1H NMR spectra. The 1H NMR spectrum for the reaction mixture of entry 12 is given in the Supplementary information file.
2-(Ethylsulfanyl)acetonitrile (4). 1H NMR spectrum (300 MHz, DMSO-d6), δ, ppm (J, Hz): 3.74 (2H, s, CH2); 2.69 (2H, q, J = 7.4, CH2); 1.24 (3H, t, J = 7.4, CH3). The spectral data coincide with those published in the literature.16
Synthesis of 3-amino-1-benzothiophene-2-carbonitriles 3a–g (General method). The corresponding 2-fluorobenzonitrile 1a–g (0.83 mmol, 1 equiv) was dissolved in DMSO (2 ml). Thereto, a solution of xanthate 2 (133 mg, 0.83 mmol, 1 equiv) in DMSO (1 ml) and DBU (529 mg, 3.47 mmol, 4.2 equiv) were added. The reaction mixture was stirred at room temperature, adding an additional portion (133 mg, 0.83 mmol, 1 equiv) of xanthate 2 every 2 h. The total amount of added xanthate 2 was 4 equiv, while the total reaction time was 8 h. Then, the reaction mixture was diluted with EtOAc (50 ml), the mixture was treated with saturated aqueous KCl solution (5×50 ml) to remove DMSO and water-soluble impurities, and the organic phase was dried over Na2SO4. The solvent was evaporated, and the product was purified by flash chromatography (eluent hexane–EtOAc, 9:1 for benzothiophenes 3a–f and 3:1 for benzothiophene 3g).
3-Amino-1-benzothiophene-2-carbonitrile (3a). Yield 144 mg (81%), gray powder, mp 157–159°C. 1H NMR spectrum (800 MHz, DMSO-d6), δ, ppm (J, Hz): 8.10 (1H, d, J = 8.1, H Ar); 7.87 (1H, d, J = 8.1, H Ar); 7.56–7.52 (1H, m, H Ar); 7.47–7.41 (1H, m, H Ar); 7.11 (2H, br. s, NH2). 13C NMR spectrum (201 MHz, DMSO-d6), δ, ppm: 152.2; 138.9; 130.2; 128.6; 124.5; 123.1; 122.8; 116.2; 72.4. Found, m/z: 175.0325 [M+H]+. C9H7N2S. Calculated, m/z: 175.0324. The spectral characteristics and melting point coincide with those published in the literature.10
3-Amino-6-fluoro-1-benzothiophene-2-carbonitrile (3b). Yield 140 mg (88%), yellow powder, mp 155–157°C. 1H NMR spectrum (800 MHz, DMSO-d6), δ, ppm (J, Hz): 8.14 (1H, dd, J = 9.0, J = 5.2, H Ar); 7.83 (1H, dd, J = 9.2, J = 2.4, H Ar); 7.35 (1H, ddd, J = 9.0, J = 9.0, J = 2.4, H Ar); 7.16 (2H, br. s, NH2). 13C NMR spectrum (201 MHz, DMSO-d6), δ, ppm (J, Hz): 162.5 (d, J = 246.6); 151.7; 140.4 (d, J = 11.3); 127.1; 124.7 (d, J = 9.7); 115.9; 113.5 (d, J = 24.5); 109.4 (d, J = 26.3); 72.4. Found, m/z: 193.0231 [M+H]+. C9H6FN2S. Calculated, m/z: 193.0230.
3-Amino-5-fluoro-1-benzothiophene-2-carbonitrile (3c). Yield 144 mg (91%), yellow powder, mp 179–181°C. 1H NMR spectrum (800 MHz, DMSO-d6), δ, ppm (J, Hz): 7.98 (1H, dd, J = 9.8, J = 2.6, H Ar); 7.94 (1H, dd, J = 8.9, J = 4.8, H Ar); 7.45 (1H, ddd, J = 8.9, J = 8.9, J = 2.6, H Ar); 7.11 (2H, br. s, NH2). 13C NMR spectrum (201 MHz, DMSO-d6), δ, ppm (J, Hz): 160.1 (d, J = 240.9); 151.6 (d, J = 4.1); 134.5; 131.3 (d, J = 9.4); 125.1 (d, J = 8.9); 117.2 (d, J = 25.1); 115.7; 108.5 (d, J = 24.2); 74.7. Found, m/z: 193.0230 [M+H]+. C9H6FN2S. Calculated, m/z: 193.0230.
3-Amino-4-fluoro-1-benzothiophene-2-carbonitrile (3d). Yield 125 mg (79%), yellow powder, mp 122–124°C. 1H NMR spectrum (800 MHz, DMSO-d6), δ, ppm (J, Hz): 7.71 (1H, d, J = 8.1, H Ar); 7.54 (1H, ddd, J = 8.1, J = 7.9, J = 4.9, H Ar); 7.21 (1H, dd, J = 11.8, J = 7.9, H Ar); 6.77 (2H, br. s, NH2). 13C NMR spectrum (201 MHz, DMSO-d6), δ, ppm (J, Hz): 158.2 (d, J = 252.7); 150.1 (d, J = 3.6); 141.3 (d, J = 4.9); 130.0 (d, J = 7.9); 119.7 (d, J = 3.9); 118.5 (d, J = 14.6); 115.3; 110.4 (d, J = 19.2); 73.7. Found, m/z: 193.0232 [M+H]+. C9H6FN2S. Calculated, m/z: 193.0230.
3-Amino-6-bromo-1-benzothiophene-2-carbonitrile (3e). Yield 188 mg (90%), yellow powder, mp 221–223°C. 1H NMR spectrum (800 MHz, DMSO-d6), δ, ppm (J, Hz): 8.21 (1H, d, J = 1.8, H Ar); 8.03 (1H, d, J = 8.6, H Ar); 7.62 (1H, dd, J = 8.6, J = 1.8, H Ar); 7.17 (2H, br. s, NH2). 13C NMR spectrum (201 MHz, DMSO-d6), δ, ppm (J, Hz): 151.7; 140.5; 129.3; 127.7; 125.6; 124.4; 122.1; 115.8; 73.2. Found, m/z: 252.9427 [M+H]+. C9H6BrN2S. Calculated, m/z: 252.9430.
3-Amino-5-bromo-1-benzothiophene-2-carbonitrile (3f). Yield 199 mg (95%), yellow powder, mp 184–186°C. 1H NMR spectrum (800 MHz, DMSO-d6), δ, ppm (J, Hz): 8.40 (1H, d, J = 2.0, H Ar); 7.87 (1H, d, J = 8.6, H Ar); 7.69 (1H, dd, J = 8.6, J = 1.9, H Ar); 7.16 (2H, br. s, NH2). 13C NMR spectrum (201 MHz, DMSO-d6), δ, ppm (J, Hz): 151.1; 137.8; 131.9; 131.1; 125.4; 125.1; 117.8; 115.6; 74.2. Found, m/z: 252.9423 [M+H]+. C9H6BrN2S. Calculated, m/z: 252.9430.
Ethyl (3-amino-2-cyano-1-benzothiophen-6-yl)glycinate (3g). Yield 68 mg (30%), brown powder, mp 135–137°C. 1H NMR spectrum (800 MHz, DMSO-d6), δ, ppm (J, Hz): 7.76 (1H, d, J = 8.8, H Ar); 6.91–6.68 (4H, m, H Ar, NH2); 6.56 (1H, br. s, NH); 4.13 (2H, q, J = 7.1, CH2); 3.98 (2H, br. s, CH2); 1.20 (3H, t, J = 7.1, CH3). 13C NMR spectrum (201 MHz, DMSO), δ, ppm: 170.7; 152.6; 149.4; 141.4; 123.2; 120.4; 117.1; 112.8; 102.1; 67.8; 60.4; 44.4; 14.1. Found, m/z: 276.0810 [M+H]+. C13H14N3O2S. Calculated, m/z: 276.0801.
Supplementary information file containing 1H, 13C NMR spectra and high-resolution mass spectra of the synthesized compounds, as well as the 1H NMR spectrum of the reaction mixture of entry 12 (Table 1) for optimizing of the synthesis conditions of 3-amino-1-benzothiophene- 2-carbonitrile (3a) is available at the journal website http://springerlink.bibliotecabuap.elogim.com/journal/10593.
References
Nagesh, H.; Padmashali, B.; Sandeep, C.; Yuvaraj, T. C. M.; Siddesh, M. B.; Mallikarjuna, S. M. Int. J. Pharm. Sci. Rev. Res. 2014, 28(2), 6.
Naganagowda, G.; Padmashali, B. Phosphorus, Sulfur Silicon Relat. Elem. 2010, 185, 1691.
Romagnoli, R.; Baraldi, P. G.; Carrion, M. D.; Cara, C. L.; Preti, D.; Fruttarolo, F.; Pavani, M. G.; Tabrizi, M. A.; Tolomeo, M.; Grimaudo, S.; Di Cristina, A.; Balzarini, J.; Hadfield, J. A.; Brancale, A.; Hamel, E. J. Med. Chem. 2007, 50, 2273.
Pinto, E.; Queiroz, M. J. R. P.; Vale-Silva, L. A.; Oliveira, J. F.; Begouin, A.; Begouin, J. M.; Kirsch, G. Bioorg. Med. Chem. 2008, 16, 8172.
Khovidhunkit, W.; Shoback, D. M. Ann. Intern. Med. 1999, 130, 431.
Saravanan, G.; Alagarsamy, V.; Chinnasamy, A.; Prakash, R. Int. J. Pharm. Pharm. Sci. 2010, 2, 8386.
Keri, R. S.; Chand, K.; Budagumpi, S.; Balappa Somappa, S.; Patil, S. A.; Nagaraja, B. M. Eur. J. Med. Chem. 2017, 138, 1002.
Beck, J. R.; Yahner, J. A. J. Org. Chem. 1974, 39, 3440.
Loidreau, Y.; Dubouilh-Benard, C.; Marchand, P.; Nourrisson, M.-R.; Duflos, M.; Buquet, C.; Corbière, C.; Besson, T. J. Heterocycl. Chem. 2013, 50, 1187.
Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.-R.; Duflos, M.; Lozach, O.; Loaëc, N.; Meijer, L.; Besson, T. Eur. J. Med. Chem. 2012, 58, 171.
Carrington, D. E. L.; Clarke, K.; Scrowston, R. M. J. Chem. Soc. C 1971, 3903.
Sokolov, A. I.; Mikhaylov, A. A.; Baleeva, N. S.; Baranov, M. S. Eur. J. Org. Chem. 2021, 4350.
Tanaka, K.; Ono, N.; Kubo, Y.; Kaji, A. Synthesis 1979, 890.
Abdellaoui, H.; Chen, X.; Xu, J. Synthesis 2017, 49, 2250.
Huang, Z.; Xu, J. RSC Adv. 2013, 3, 15114.
https://scifinder-n.cas.org/searchDetail/substance/655e066225b43e48fc8741ae/substanceSpectra
The study was carried out with financial support from the Russian Science Foundation within the framework of scientific project No. 20-73-10195.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2024, 60(1/2), 99–102
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ivanov, D.S., Sokolov, A.I., Smirnov, A.Y. et al. A novel approach to obtain 3-amino-1-benzothiophene-2-carbonitriles. Chem Heterocycl Comp 60, 99–102 (2024). https://doi.org/10.1007/s10593-024-03299-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-024-03299-y