

Synthesis of amino acids with isolated saturated (hetero)carbocyclic scaffold was disclosed. The method relied on Suzuki cross-coupling reaction of heteroacycloalkene triflates and N-Boc-tetrahydropyridine boron pinacolates followed by catalytic hydrogenation of the conjugated dienes thus obtained, which proceeded with good diastereoselectivity for scaffolds with two six-membered cycles and low diastereoselectivity for a five-membered analog. Several amino acid derivatives have also been obtained, i.e. amino esters, NH- and N-Boc-substituted amino acids – promising building blocks for medicinal and synthetic chemistry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
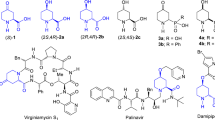
Synthetic amino acids,1,2 especially cyclic derivatives,3,4,5,6 play an important role in medicinal chemistry and drug discovery. Meanwhile, the high demand for new scaffolds is the driving force of modern chemistry design concepts and research programs. Latest promising three-dimensional scaffolds bear sp3-enriched saturated isolated (hetero)-carbocycles (Fig. 1). The most prominent examples of compounds with the latter type of connectivity included rociverine,7,8 dicyclomine (dicycloverine),9 benzetimide,10,11,12 or fosinopril (monopril).13,14,15

Pharmaceutically relevant examples of compounds with isolated (hetero)aliphatic rings scaffold.
However, the synthetic accessibility of these scaffolds is limited because formal C(sp3)–C(sp3) coupling16 is required to connect two cycles, which is a difficult task to achieve by most synthetic methods. Limited examples included the silyl-mediated photoredox Giese reaction (addition of nonactivated alkyl bromides to alkenes) in presence of Ir[dF(CF3)ppy]2(dtbbpy)PF6 (Scheme 1a)17 or Lewis acid promoted C(sp3)–C(sp3) coupling reaction of organozinc compounds18 (Scheme 1b).

Synthetic approaches to amino esters with isolated (hetero)aliphatic rings
The construction of the target ring systems can be performed via the indirect approach involving a conventional C(sp2)–C(sp2) coupling followed by the reduction of both double bonds of dienes thus formed. Following this strategy, we have aimed at the synthesis of amino acid derivatives derived from isolated-ring scaffolds including cyclopentane and cyclohexane rings, as well as tetrahydropyranyl and piperidinyl moieties via the cross-coupling reaction of (hetero)cycloalkenes followed by the catalytic hydrogenation (Scheme 1c). It was envisaged that the presence of two functional groups could be used to obtain various derivatives of mono- or deprotected amino acids.
First, commercially available α-oxo esters 1a–d were subjected to the reaction with PhN(OTf)2 and NaH19 (Scheme 2). This approach led to preparation of cycloalkenyl triflates 2a–d in good to high yields (78–90%). These compounds were used in the subsequent Suzuki cross-coupling reaction20 with N-Boc-tetrahydropyridinyl-4-boron pinacolate (3). All reactions were carried out using Pd(dppf)Cl2 as a catalyst and K2CO3 as a base in 1,4-dioxane–H2O at 100°C, which allowed for the preparation of diene carboxylates 4a–d in 56–70% yield after chromatographic purification (Scheme 2).

Synthesis of dienes 4a–d using Suzuki reaction of (hetero)cycloalkenyl triflates 2a–d with tetrahydropyridine boronate 3
The key step of the reaction sequence included the reduction of obtained dienes 4a–d to the corresponding N-Boc-amino esters 5a–d (Scheme 3). The hydrogenation of dienes to achieve formal C(sp3)–C(sp3) coupling connectivity appeared to be a challenging task due to the stability of conjugated polysubstituted diene carboxylate fragments. Thus, catalytic hydrogenation of compounds 4a,b was recently studied and optimized by our group by testing a series of Pd/C and Pd(OH)2/C catalysts.19 It was found that a Pd/C composite prepared by deposition of Pd nanoparticles (5–40 nm) via the reduction of Pd2+ with formaldehyde20 demonstrated the best results in such transformations.

Reduction of dienes 4a–d to saturated compounds 5a–d (relative configurations are shown)
This catalyst was applied to a series of diene carboxylates 4a–d. The hydrogenation proceeded in an autoclave under 100 atm of H2 at 30°C for 72 h to ensure the complete conversion and reduction of both C=C double bonds. The reaction proceeded in a diastereoselective manner in the case of scaffolds containing two six-membered rings, i.e., for compounds 4b–d. The corresponding saturated amino esters were obtained as a ca. 9:2 mixture of cis- and transdiastereomers (for compound 5b) or cis-isomers as sole products (for compounds 5c,d). Cyclopentane carboxylate 5a was obtained with dr 8:7 that retained after chromatographic purification.
N-Boc-amino esters 5a–c could be easily transformed into other valuable amino acid-derived building blocks (Scheme 4). In particular, alkaline hydrolysis of the ester groups of derivatives 5a–c was successfully achieved by refluxing with NaOH in EtOH–H2O followed by acidification with aq NaHSO4, leading to N-Boc-amino acids 6a–c. Derivatives 6a–c were obtained in good yields, however significant epimerization was observed, so that the products were obtained as ca. 1:1 mixture of cis- and transdiastereomers. The dr ratio could be changed after the chromatographic purification used to obtain analytically pure samples according to 2D NMR experiments. Variation of the reaction conditions also resulted in epimerization, but in addition gave significantly lower yield of the target products 6a–c. At the same time, N-Boc deprotection of the compounds 5a–c in 1,4-dioxane–HCl at room temperature proceeded without epimerization and gave amino esters 7a–c in high yield (89–95%).

Synthesis of amino acids and their derivatives from compounds 5a–c (relative configurations are shown)
Hydrolysis of ester group of esters 7a–c proceeded in the way similar to the case of synthesis of compounds 6a–c and provided title amino acids 8a–c in 45–92% yield. The possibility of obtaining salt forms based on both the amino and the carboxyl groups was revealed. All final products were purified by HPLC. The structures and dr values of the products, as well as the relative configurations, were confirmed by a series of NMR experiments (see Supplementary information file for details). Attempted acidic hydrolysis of both ester and carbamate groups in molecules 5a–c was inefficient and resulted in low yields of the target products 8a–c. Finally, the KOH-mediated alkaline hydrolysis of bispiperidine derivative 5d also proceeded with epimerization, and product 9 was isolated as a single diastereomer in 40% yield (Scheme 5).

Synthesis of potassium 1′-(tert-butoxycarbonyl)-[3,4′-bipiperidine]-4-carboxylate (9) (relative configurations are shown)
In conclusion, we have developed a straightforward and robust approach to amino acid derivatives based on scaffolds with two isolated saturated (hetero)carbocycles. The method relied on the C(sp2)–C(sp2) Suzuki cross-coupling reaction of heterocyclic alkenyl triflates and N-Boc-tetrahydropyridine- derived boronate.
We found suitable catalysts for the key step – catalytic hydrogenation of the conjugated dienes to achieve C(sp3)–C(sp3) connectivity, which proceeded with good diastereoselectivity for scaffolds with two six-membered cycles, and with dr ratio of ca. 1:1 for a five-membered counterpart. The resulting N-Boc-amino esters are convenient reagents for the preparation of free amino acids and their monoprotected derivatives. Notably, the alkaline hydrolysis reaction of the ester groups appeared to be somewhat challenging: while it gave good yields of the target products, it was accompanied by epimerization. Though we did not check applicability of the catalysts, proposed herein, for a wider range of dienes, we suppose that the reported procedure can be helpful for preparation of a series of similar compounds.
In our opinion, the proposed method to obtain scaffolds with saturated isolated cycles could be useful for synthetic chemists, while the synthesized compounds could be considered as promising building blocks21 for the purposes of medicinal chemistry and drug discovery.
Experimental
1H and 13C NMR spectra were recorded on an Agilent ProPulse 600 spectrometer (600 and 151 MHz, respectively), a Bruker 170 Avance 500 spectrometer (500 and 126 MHz, respectively), and a Varian Unity Plus 400 spectrometer (400 and 101 MHz, respectively). Internal standards: TMS and residual solvent signals at 7.26 and 77.2 ppm for 1H and 13C nuclei in CDCl3, 2.50 and 39.5 ppm for 1H and 13C nuclei in DMSO-d6. Mass spectra were recorded on an Agilent 1100 LCMSD SL instrument (chemical ionization (CI)) and an Agilent 5890 Series II 5972 GCMS instrument (electron impact ionization (EI)). Elemental analyses were performed at the Laboratory of Organic Analysis, Department of Chemistry, Taras Shevchenko National University of Kyiv. The reaction process was monitored by 1H NMR and mass spectroscopy. Melting points were measured on a MPA100 OptiMelt automated melting point system. Analytical TLC was performed using Polychrom SI F254 plates. Column chromatography was performed using silica gel (230–400 mesh) as the stationary phase.
The solvents were purified according to the standard procedures.22 Compounds 1a–d and 3 were available from Enamine Ltd.
Preparation of alkenyl triflates 2a–d23,24,25,26 (General method). 0.2 M Solution of the corresponding α-keto ester (57.0 mmol) in THF (285 ml) was cooled to 0°C, and NaH (60% in mineral oil, 2.30 g, 57.5 mmol) was added in portions. The reaction mixture was stirred at 0°C for 30 min, then PhN(OTf)2 (20.5 g, 57.5 mmol) was added. The resulting mixture was stirred at room temperature overnight, then poured into H2O (500 ml) and extracted with EtOAc (2×300 ml). Combined organic layers were washed with brine (2×100 ml), dried over Na2SO4, filtered, and evaporated in vacuo to dryness. The crude product was purified by column chromatography on silica gel using hexanes–EtOAc, 20:1 as eluent.
Synthesis of diene carboxylates 4a–d by Suzuki cross-coupling reaction (General method). The corresponding triflate 2a–d (3.65 mmol) was dissolved in 1,4-dioxane–H2O (14 ml, 7:2), then K2CO3 (1.51 g, 10.9 mmol), Pd(dppf)Cl2 (267 mg, 0.365 mmol), and tert-butyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3,6-dihydropyridine-1(2H)-carboxylate (3) (1.24 g, 4.01 mmol) were added. The reactor was degassed, purged with argon, and the reaction mixture was stirred at 100°C for 16 h. The resulting mixture was cooled to room temperature, EtOAc (40 ml) was added, the resulting solution was washed with H2O (3×15 ml) and brine (3×15 ml), dried over Na2SO4, filtered, and evaporated in vacuo to dryness.
tert-Butyl 4-[2-(methoxycarbonyl)cyclopent-1-en-1-yl]-3,6-dihydropyridine-1(2H)-carboxylate (4a). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. Yield 729 mg (65%), yellowish oil. The compound existed as a ca. 1:1 mixture of rotamers. 1H NMR spectrum (400 MHz, CDCl3), δ, ppm (J, Hz): 5.91–5.38 (1H, m, H-4); 4.02–3.91 (2H, m, H-5); 3.68 (3H, s, CO2CH3); 3.48 (2H, t, J = 5.6, H-1); 2.67 (2H, t, J = 7.6, H-2); 2.59 (2H, td, J = 7.6, J = 2.3, H-12); 2.29–2.17 (2H, m, H-14); 1.85 (2H, quint, J = 7.6, H-13); 1.44 (9H, s, C(CH3)3). 13C NMR spectrum (126 MHz, CDCl3), δ, ppm: 166.7; 154.3; 152.0; 132.8; 127.6; 122.9; 122.3; 79.1; 50.9; 43.2; 42.6; 40.5; 39.2; 37.7; 34.7; 28.0; 27.2; 21.2. Mass spectrum (ES-API), m/z: 208 [M–CH=C(CH3)2–CO2+H]+. Found, %: C 66.11; H 7.85; N 4.57. C17H25NO4. Calculated, %: C 66.43; H 8.20; N 4.56.
tert-Butyl 4-[2-(ethoxycarbonyl)cyclohex-1-en-1-yl]-3,6-dihydropyridine-1(2H)-carboxylate (4b). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. Yield 747 mg (61%), yellowish oil. The compound existed as a ca. 3:2 mixture of rotamers. 1H NMR spectrum (400 MHz, CDCl3), δ, ppm (J, Hz): 5.38–5.19 (1H, m, H-4); 4.11 (2H, q, J = 7.1, H-19); 3.93–3.81 (2H, m, H-5); 3.53 (2H, t, J = 5.5, H-1); 2.29 (2H, dt, J = 6.6, J = 3.7, H-2); 2.24–2.16 (2H, m, H-12); 2.13 (2H, dd, J = 6.6, J = 3.7, H-15); 1.68–1.57 (4H, m, H-13,14); 1.47 (9H, s, C(CH3)3); 1.22 (3H, t, J = 7.1, H-20). 13C NMR spectrum (126 MHz, CDCl3), δ, ppm: 169.2; 154.4; 154.3; 146.2; 146.0; 138.9; 138.7; 125.8; 118.2; 117.9; 79.0; 59.6; 42.9; 42.4; 40.4; 39.2; 29.6; 28.0; 27.3; 27.2; 25.7; 21.6; 21.5; 13.8. Mass spectrum (ES-API), m/z: 236 [M–CH=C(CH3)2–CO2+H]+. Found, %: C 68.16; H 9.06; N 4.51. C19H29NO4. Calculated, %: C 68.03; H 8.71; N 4.18.
tert-Butyl 4-[5-(methoxycarbonyl)-3,6-dihydro-2Hpyran-4-yl]-3,6-dihydropyridine-1(2H)-carboxylate (4c). The product was purified by flash column chromatography (5 bar, 80 g column) using CHCl3–EtOAc as eluent, gradient from 1:0 to 0:1. Yield 826 mg (70%), colorless oil. The compound existed as a ca. 1:1 mixture of rotamers. 1H NMR spectrum (400 MHz, CDCl3), δ, ppm (J, Hz): 5.48–5.30 (1H, m, H-4); 4.32 (2H, t, J = 2.5, H-14); 4.00–3.88 (2H, m, H-5); 3.78 (2H, t, J = 5.6, H-10); 3.68 (3H, s, CO2CH3); 3.57 (2H, t, J = 5.6, H-1); 2.35–2.26 (2H, m, H-11); 2.25–2.14 (2H, m, H-2); 1.48 (9H, s, C(CH3)3). 13C NMR spectrum (126 MHz, CDCl3), δ, ppm: 166.2; 155.0; 148.3; 138.1; 124.4; 119.5; 79.8; 65.5; 64.0; 51.5; 43.6; 43.1; 41.3; 40.1; 31.0; 28.6; 27.9. Mass spectrum (ES-API), m/z: 224 [M–CH=C(CH3)2–CO2+H]+, 346 [M+Na]+. Found, %: C 63.45; H 8.19; N 4.50. C17H25NO5. Calculated, %: C 63.14; H 7.79; N 4.33.
1'-(tert-Butyl) 4-ethyl 1-benzyl-1,2,3',5,6,6'-hexahydro-[3,4'-bipyridine]-1',4(2'H)-dicarboxylate (4d). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. Yield 874 mg (56%), yellowish oil. The compound existed as a ca. 11:9 mixture of rotamers. 1H NMR spectrum (400 MHz, CDCl3), δ, ppm (J, Hz): 7.47–7.27 (5H, m, H Ph); 5.45–5.35 (1H, m, H-10); 5.15 (2H, s, CH2Ph); 4.14 (2H, q, J = 7.1, H-26); 4.03–3.96 (2H, m, H-11); 3.94–3.87 (2H, m, H-1); 3.61–3.51 (4H, m, H-5,7); 2.48–2.39 (2H, m, H-4); 2.25–2.16 (2H, m, H-8); 1.47 (9H, s, C(CH3)3); 1.24 (3H, t, J = 7.1, H-27). 13C NMR spectrum (151 MHz, CDCl3, some signals are doubled due to the presence of rotamers), δ, ppm: 167.5; 155.2; 154.9; 136.7; 134.2; 128.7; 128.3; 128.2; 124.9; 124.5; 121.6; 121.1; 79.8; 67.4; 60.7; 47.2; 43.6; 43.0; 41.0; 40.5; 40.2; 39.8; 28.6; 28.3; 26.1; 25.7; 14.4. Mass spectrum (ES-API), m/z: 371 [M–CH=C(CH3)2+H]+. Found, %: C 70.45; H 8.35; N 6.73. C25H34N2O4. Calculated, %: C 70.40; H 8.03; N 6.57.
Synthesis of N-Boc-amino esters 5a–d (General method). Compound 4a–d (20.0 mmol), the optimal nanocomposite 10% Pd/C20 (10 mol % counting per Pd), and MeOH (140 ml) were mixed directly in an autoclave, which was purged with argon. The autoclave was sealed, purged with hydrogen, and pressurized with hydrogen quickly after the mixing of the reagents. Hydrogenation was carried out under 100 atm of H2 at 30°C for 72 h (NOTE: as it was found, this time is required to achieve the complete hydrogenation in most cases). Then, the catalyst was separated by centrifugation, the reaction mixture was evaporated, and the organic residue was analyzed by NMR and LC/MS.
tert-Butyl 4-[2-(methoxycarbonyl)cyclopentyl]piperidine-1-carboxylate (5a). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. The compound was obtained as a ca. 8:7 mixture of diastereomers. Yield 5.59 g (90%), yellowish oil. 1H NMR spectrum (400 MHz, CDCl3), δ, ppm (J, Hz): 4.05 (2H, t, J = 12.9, H-2,6); 3.64 (1.4H, s, CO2CH3); 3.62 (1.6H, s, CO2CH3); 2.90 (0.53H, t, J = 7.1, H-11); 2.51–2.36 (0.47H, m, H-11); 2.67–2.54 (2H, m, H-2,6); 2.08–1.99 (0.47H, m, CH, CH2); 1.94–1.69 (4.53H, m, H-3–5,10,12–14); 1.68–1.45 (5H, m, H-3–5,10,12–14); 1.42 (9H, s, C(CH3)3); 1.39–1.17 (1.87H, m, CH, CH2); 1.16–1.01 (2.13H, m, CH, CH2). 13C NMR spectrum (126 MHz, CDCl3), δ, ppm: 177.1; 175.8; 154.4; 154.3; 78.8; 78.7; 51.2; 50.3; 50.6; 47.0; 48.6; 45.2; 43.6; 43.5 (2C); 43.4; 40.4; 37.6; 31.3; 31.1; 30.9; 30.3; 29.8; 29.5; 29.1; 28.0; 24.9; 22.8. Mass spectrum (ES-API), m/z: 312 [M+H]+. Found, %: C 65.40; H 9.53; N 4.10. C17H29NO4. Calculated, %: C 65.57; H 9.39; N 4.50.
cis-tert-Butyl 4-[2-(ethoxycarbonyl)cyclohexyl]piperidine-1-carboxylate (5b). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. The compound was obtained as a single cis-diastereomer. Yield 6.29 g (93%), yellowish oil. 1H NMR spectrum (400 MHz, CDCl3), δ, ppm (J, Hz): 4.19–3.97 (4H, m, H-2,6,19); 2.83–2.72 (1H, m, H-11); 2.65–2.52 (2H, m, H-2,6); 2.02–1.90 (1H, m, H-12); 1.83–1.69 (3H, m, H-3,5,13); 1.67–1.55 (3H, m, H-12,14,15); 1.50–1.45 (3H, m, H-4,12,14); 1.42 (9H, s, (C(CH3)3); 1.22 (3H, t, J = 7.1, H-20); 1.20–1.09 (2H, m, H-10,13); 1.04–0.90 (2H, m, H-3,5). 13C NMR spectrum (126 MHz, CDCl3), δ, ppm: 174.1; 154.3; 78.7; 59.2; 43.9; 43.5; 40.6; 37.8; 29.9; 29.6; 28.8; 28.0; 25.5; 24.6; 21.5; 13.8. Mass spectrum (ES-API), m/z: 340 [M+H]+. Found, %: C 66.88; H 10.09; N 4.28. C19H33NO4. Calculated, %: C 67.22; H 9.80; N 4.13.
cis-tert-Butyl 4-[3-(methoxycarbonyl)tetrahydro-2Hpyran-4-yl]piperidine-1-carboxylate (5c). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. The compound was obtained as a single cis-diastereomer. Yield 3.92 g (60%), yellowish oil. 1H NMR spectrum (400 MHz, CDCl3), δ, ppm (J, Hz): 4.25 (1H, d, J = 11.7, H-10,14); 4.19–3.96 (3H, m, H-2,6,10,14); 3.70 (3H, s, CO2CH3); 3.50 (1H, dd, J = 11.7, J = 3.1, H-10,14); 3.38 (1H, td, J = 12.0, J = 2.6, H-11); 2.74–2.49 (3H, m, H-2,6); 2.11–1.98 (1H, m, H-12); 1.87–1.76 (1H, m, H-3–5,13); 1.72–1.61 (2H, m, H-3–5,13); 1.59–1.53 (1H, m, H-3–5,13); 1.49–1.32 (10H, m, C(CH3)3, H-3-5,13); 1.15–0.83 (2H, m, H-3–5,13). 13C NMR spectrum (101 MHz, CDCl3), δ, ppm: 173.0; 154.9; 79.4; 70.5; 69.0; 51.7; 43.9; 42.6; 42.0; 37.7; 30.2; 29.7; 28.6; 26.0. Mass spectrum (ES-API), m/z: 228 [M–H2C=C(CH3)2–CO2+H]+. Found, %: C 62.31; H 8.94; N 3.98. C17H29NO5. Calculated, %: C 62.36; H 8.93; N 4.28.
cis-3-[1-(tert-Butoxycarbonyl)piperidin-4-yl]-4-(ethoxycarbonyl)piperidin-1-ium chloride (5d). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. After that, the amine (ca. 20.0 mmol) and i-Pr2EtN (3.48 ml, 20.0 mmol) were dissolved in CH2Cl2 (200 ml) and benzyl chloroformate (2.85 ml, 20.0 mmol) was added at room temperature. After 1 h, saturated aq NaHCO3 (500 ml) was added and the aqueous phase was extracted with EtOAc (3×100 ml). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by flash chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1.23 The obtained N-Cbz-diamine (ca. 20.0 mmol) was dissolved in MeOH (75 ml). Pd/C (0.75 g) and CHCl3 (1.60 ml, 20.0 mmol) were added. The mixture was hydrogenated under H2 (1 bar) at 40°C for 16 h, then catalyst was filtered off, and filtrate was evaporated in vacuo. The residue was washed with EtOAc (3×25 ml) and dried in air.5 The compound was obtained as a single cis-diastereomer. Overall yield 5.71 g (76%), colorless powder, mp 159–160°C. 1H NMR spectrum (500 MHz, CDCl3), δ, ppm (J, Hz): 9.73 (1H, d, J = 9.9, NH2+); 9.46 (1H, d, J = 9.9, NH2+); 4.25–4.04 (4H, m, H-10,13,17); 3.40–3.16 (4H, m, H-2,6,13,17); 2.97–2.91 (1H, m, H-2,6); 2.65–2.52 (2H, m, H-2,4,6); 2.30–2.19 (1H, m, H-5); 2.09–2.02 (1H, m, H-5); 2.02–1.94 (1H, m, H-3); 1.78–1.71 (1H, m, H-14–16); 1.68–1.61 (1H, m, H-14–16); 1.52–1.32 (10H, m, C(CH3)3, H-14–16); 1.25 (3H, t, J = 7.1, H-11); 1.21–1.06 (2H, m, H-14–16). 13C NMR spectrum (126 MHz, CDCl3), δ, ppm: 172.9; 154.7; 79.7; 61.0; 43.6; 42.6; 40.1; 39.8; 37.5; 36.5; 29.8; 29.3; 28.6; 25.3; 14.4. Mass spectrum (ES-API), m/z: 341 [M–HCl+H]+. Found, %: C 57.46; H 8.62; N 7.08; Cl 9.14. C18H33ClN2O4. Calculated, %: C 57.36; H 8.83; N 7.43; Cl 9.41.
Synthesis of N-Boc-amino acids 6a–c (General method). Synthesized crude compound 5a–c (5.00 mmol) was added to NaOH (400 mg, 10.0 mmol) in EtOH–H2O (30 ml, 7:3), and the resulting mixture was refluxed for 16 h. Then, reaction mixture was cooled to room temperature, and byproducts were extracted with t-BuOMe (2×10 ml). Then, saturated aq NaHSO4 was added until pH 5 was adjusted. After that, aqueous solution was extracted with t-BuOMe (3×10 ml). Combined organic layers were dried over Na2SO4 and evaporated in vacuo.
2-[1-(tert-Butoxycarbonyl)piperidin-4-yl]cyclopentane-1-carboxylic acid (6a). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. The compound was obtained as a 3:7 mixture of cis-/trans-diastereomers; existed as a mixture of rotamers. Yield 993 mg (67%), yellowish oil. 1H NMR spectrum (500 MHz, CDCl3), δ, ppm (J, Hz): 8.81 (1H, br. s, CO2H); 4.21–3.98 (2H, m, H-10,14); 3.56–3.48 (0.3H, m, H-1); 2.93 (0.3H, t, J = 6.8, H-1); 2.78–2.60 (2.4H, m, H-1,10,14); 2.56–2.46 (1H, m, H-2); 2.14–1.76 (4H, m, H-3,5,11,13); 1.75–1.49 (4H, m, H-3,5,11,13); 1.48–1.05 (12H, m, C(CH3)3, H-4,12). 13C NMR spectrum (126 MHz, CDCl3), δ, ppm: 182.8; 181.4; 171.0; 165.5; 155.0; 126.7; 79.6; 79.5 (2C); 51.0; 49.2; 47.5; 45.8; 44.1; 41.0; 38.0; 36.9; 34.8; 33.5; 31.9; 31.7; 31.6; 31.0; 30.5; 30.1; 30.0; 29.8; 28.6; 28.5; 25.7; 23.3; 21.5. Mass spectrum (ES-API), m/z: 242 [M–CH=C(CH3)2+H]+, 320 [M+Na]+. Found, %: C 64.69; H 9.11; N 4.36. C16H27NO4. Calculated, %: C 64.62; H 9.15; N 4.71.
2-[1-(tert-Butoxycarbonyl)piperidin-4-yl]cyclohexane-1-carboxylic acid (6b). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. The compound was obtained as a 3:2 mixture of cis-/trans-diastereomers. Yield 1.24 g (80%), colorless solid, mp 134–135°C. 1H NMR spectrum (600 MHz, DMSO-d6), δ, ppm (J, Hz): 11.95 (1H, s, CO2H); 4.03–3.86 (2H, m, H-11,15); 2.75–2.49 (3H, m, H-1,11,15); 2.13 (0.4H, td, J = 11.3, J = 3.5, H-2); 1.91–1.86 (0.6H, m, H-2); 1.82–1.77 (0.4H, m, H-13); 1.77–1.73 (0.6H, m, H-13); 1.71–1.61 (2H, m, H-6); 1.58–1.21 (15H, m, C(CH3)3, H-4,5,12,14); 1.21–1.11 (2H, m, H-4,5); 1.05–0.81 (2H, m, H-3). 13C NMR spectrum (126 MHz, CDCl3, some signals are doubled due to the presence of rotamers and diastereomers), δ, ppm: 181.5; 180.3; 155.0; 79.5 (2C); 47.0; 44.6; 44.5; 44.2; 43.6; 41.1; 39.2; 38.2; 30.6; 30.5; 30.4; 30.2; 29.5; 28.6; 26.3; 26.1; 25.9; 25.7; 25.6; 25.1; 22.2. Mass spectrum (ES-API), m/z: 212 [M–CO2–CH=C(CH3)2+H]+, 256 [M–CH=C(CH3)2+H]+, 334 [M+Na]+. Found, %: C 65.35; H 9.29; N 4.41. C17H29NO4. Calculated, %: C 65.57; H 9.39; N 4.50.
4-[1-(tert-Butoxycarbonyl)piperidin-4-yl]tetrahydro-2H-pyran-3-carboxylic acid (6c). The product was purified by column chromatography on silica gel using hexanes–t-BuOMe as eluent, gradient from 1:0 to 0:1. The compound was obtained as a 1:1 mixture of cis-/trans-diastereomers. Yield 1.29 g (82%), beige powder, mp 138–139°C. 1H NMR spectrum (500 MHz, CDCl3), δ, ppm (J, Hz): 9.87 (1H, br. s, CO2H); 4.33 (0.5H, d, J = 11.8, H-1,5); 4.22–4.04 (3H, m, H-1,5,11,15); 4.02–3.97 (0.5H, m, H-1,5); 3.53 (0.5H, dd, J = 11.8, J = 3.0, H-1,5); 3.48–3.36 (1.5H, m, H-1,5); 2.73–2.55 (3H, m, H-2,11,15); 1.98–1.79 (2H, m, H-2,13); 1.70–1.55 (2H, m, H-12,14); 1.51–1.30 (11H, m, C(CH3)3, H-12,14); 1.25–0.93 (2H, m, H-4). 13C NMR spectrum (126 MHz, CDCl3, some signals are doubled due to the presence of rotamers and diastereomers), δ, ppm: 177.6; 177.0; 155.0; 79.7; 79.6; 70.3; 69.5; 69.1; 68.3; 46.1; 44.3; 44.0; 42.5; 42.0; 41.6; 38.8; 37.6; 30.1; 29.8; 29.7; 28.6; 26.6; 25.9; 25.7. Mass spectrum (ES-API), m/z: 214 [M–CO2–CH=C(CH3)2+H]+, 258 [M–CH=C(CH3)2+H]+, 336 [M+Na]+. Found, %: C 61.15; H 8.99; N 4.75. C16H27NO5. Calculated, %: C 61.32; H 8.68; N 4.47.
Synthesis of amino esters 7a–c (General method). Synthesized compound 5a–c (5.00 mmol) was dissolved in 2 M HCl solution in 1,4-dioxane (30 ml), and the resulting mixture was stirred at room temperature for 16 h. The formed solid product was filtered off and washed with t-BuOMe (2×10 ml).
4-[2-(Methoxycarbonyl)cyclopentyl]piperidin-1-ium chloride (7a) was obtained as a 2:3 mixture of cis-/transdiastereomers. Yield 1.18 g (95%), beige powder, mp 100–101°C. 1H NMR spectrum (500 MHz, DMSO-d6), δ, ppm (J, Hz): 9.15 (1H, br. s, NH2+); 8.95 (1H, br. s, NH2+); 3.59 (1.8H, s, CO2CH3); 3.58 (1.2H, s, CO2CH3); 3.23–3.14 (2H, m, H-2,6); 2.91–2.86 (0.4H, m, H-8); 2.81–2.68 (2H, m, H-2,6); 2.49–2.45 (0.6H, m, H-8); 1.98–1.91 (0.6H, m, H-7); 1.90–1.64 (6H, m, H-7, CH, CH2); 1.61–1.52 (1.6H, m, CH, CH2); 1.47–1.33 (3.2H, m, CH, CH2); 1.29–1.21 (0.6H, m, CH, CH2). 13C NMR spectrum (126 MHz, DMSO-d6), δ, ppm: 176.4; 175.2; 51.5; 51.1; 49.5; 48.0; 46.5; 44.9; 44.0; 42.9; 42.8; 42.7; 37.7; 35.2; 30.9; 29.6; 28.9; 27.8; 27.8; 27.7; 27.0; 26.3; 24.9; 22.8. Mass spectrum (ES-API), m/z: 212 [M–HCl+H]+. Found, %: C 57.97; H 8.58; N 5.27; Cl 14.43. C12H22ClNO2. Calculated, %: C 58.17; H 8.95; N 5.65; Cl 14.31.
cis-4-[2-(Ethoxycarbonyl)cyclohexyl]piperidin-1-ium chloride (7b) was obtained as a single cis-diastereomer. Yield 1.22 g (89%), colorless powder, mp 149–150°C. 1H NMR spectrum (500 MHz, DMSO-d6), δ, ppm (J, Hz): 9.20–8.92 (2H, m, NH2+); 4.10–3.99 (2H, m, H-16); 3.20 (2H, t, J = 10.3, H-2,6); 2.82–2.62 (3H, m, H-2,6,8); 1.93–1.84 (2H, m, H-3–5,7,9–12); 1.83–1.76 (1H, m, H-3–5,9–12); 1.73–1.67 (1H, m, H-3–5,9–12); 1.63–1.57 (1H, m, H-3–5,9–12); 1.55–1.13 (12H, m, H-3–5,9–12,17). 13C NMR spectrum (126 MHz, DMSO-d6), δ, ppm: 173.5; 59.3; 43.2; 43.0; 42.9; 40.2; 35.6; 28.5; 26.5; 26.2; 25.3; 24.4; 21.5; 14.1. Mass spectrum (ES-API), m/z: 240 [M–HCl+H]+. Found, %: C 60.86; H 9.31; N 5.01; Cl 12.55. C14H26ClNO2. Calculated, %: C 60.97; H 9.50; N 5.08; Cl 12.85.
cis-4-[3-(Methoxycarbonyl)tetrahydro-2H-pyran-4-yl]-piperidin-1-ium chloride (7c) was obtained as a single cisdiastereomer. Yield 1.19 g (90%), beige powder, mp 206–207°C. 1H NMR spectrum (500 MHz, DMSO-d6), δ, ppm (J, Hz): 9.09 (2H, br. s, NH2+); 4.06 (1H, d, J = 11.5, H-7,11); 3.89 (1H, dd, J = 11.5, J = 4.6, H-7,11); 3.60 (3H, s, CO2CH3); 3.46 (1H, dd, J = 11.5, J = 3.0, H-7,11); 3.33–3.26 (1H, m, H-7,11); 3.23–3.18 (2H, m, H-2,6); 2.83–2.67 (3H, m, H-2,6,8); 1.94–1.86 (1H, m, H-9); 1.82–1.64 (3H, m, H-3–5,10); 1.52–1.46 (1H, m, H-3–5,10); 1.44–1.23 (3H, m, H-3–5,10). 13C NMR spectrum (126 MHz, DMSO-d6), δ, ppm: 172.2; 69.5; 67.7; 51.2; 42.9; 42.8; 41.0; 40.9; 34.9; 26.2; 25.7; 25.2. Mass spectrum (ES-API), m/z: 228 [M–HCl+H]+. Found, %: C 54.86; H 8.03; N 5.27; Cl 13.35. C12H22ClNO3. Calculated, %: C 54.65; H 8.41; N 5.31; Cl 13.44.
Synthesis of amino acids 8a–c (General method). Synthesized crude compound 7a–c (3.00 mmol) was added to NaOH (240 mg, 6.00 mmol for compounds 7a,b) or KOH (336 mg, 6.00 mmol for compound 7c) in EtOH–H2O (18 ml, 7:3), and the resulting mixture was refluxed for 16 h. Then, reaction mixture was cooled to room temperature, and evaporated to dryness.
4-(2-Carboxycyclopentyl)piperidin-1-ium chloride (8a). Then 2 M HCl–1,4-dioxane (10 ml) was added to the obtained compound, and the resulting mixture was stirred at room temperature for 15 min. After that, the precipitate was filtered off, and the target product was purified by HPLC using H2O–MeCN as eluent, gradient from 1:0 to 0:1 (30 ml/min flow, XBridge BEH C18 column). The compound was obtained as a 1:4 mixture of cis- and trans-diastereomers. Yield 342 mg (49%), brownish powder, mp 183–184°C. 1H NMR spectrum (500 MHz, DMSO-d6), δ, ppm (J, Hz): 12.09 (1H, br. s, CO2H); 9.24–9.03 (1H, m, NH2+); 8.98–8.81 (1H, m, NH2+); 3.25–3.13 (2H, m, H-2,6); 2.84–2.67 (2H, m, H-2,6); 2.38 (1H, q, J = 7.9, H-8); 1.99–1.89 (1H, m, H-7); 1.88–1.62 (5H, m, H-3,5,9,10,11); 1.60–1.32 (5H, m, H-3–5,10,11); 1.28–1.18 (1H, m, H-3,5,10,11). 13C NMR spectrum (126 MHz, DMSO-d6, some signals are doubled due to the presence of diastereomers), δ, ppm: 177.6; 176.4; 49.4; 47.7; 46.9; 45.1; 43.0; 43.0; 42.9; 42.8; 37.9; 35.2; 31.0; 29.7; 29.1; 27.9; 27.8 (2C); 27.1; 26.4; 25.1; 22.8. Mass spectrum (ES-API), m/z: 198 [M–HCl+H]+. Found, %: C 56.76; H 8.89; N 6.12; Cl 15.39. C11H20ClNO2. Calculated, %: C 56.52; H 8.62; N 5.99; Cl 15.17.
4-(2-Carboxycyclohexyl)piperidin-1-ium chloride (8b). 2 M HCl–1,4-dioxane (10 ml) was added to the obtained compound, and the resulting mixture was stirred at room temperature for 15 min. After that, the precipitate was filtered off, and the target product was purified by HPLC using H2O–MeCN as eluent, gradient from 1:0 to 0:1 (30 ml/min flow, XBridge BEH C18 column). The compound was obtained as a 1:1 mixture of cis- and trans-diastereomers. Yield 684 mg (92%), yellowish powder, mp 184–185°C. 1H NMR spectrum (500 MHz, DMSO-d6), δ, ppm (J, Hz): 12.08 (1H, br. s, CO2H); 9.19–8.80 (2H, m, NH2+); 3.28–3.16 (2H, m, H-2,6); 2.86–2.74 (1H, m, H-2,6); 2.74–2.59 (1.5H, m, H-2,6,8); 2.16 (0.5H, td, J = 11.1, J = 3.5, H-8); 1.97–1.86 (1H, m, CH, CH2); 1.85–1.79 (1H, m, CH, CH2); 1.72–1.14 (11.5H, m, CH, CH2); 1.01–0.93 (0.5H, m, CH, CH2). 13C NMR spectrum (126 MHz, DMSO-d6, some signals are doubled due to the presence of diastereomers), δ, ppm: 176.5; 175.3; 46.2; 43.5; 43.3; 43.1; 43.0; 42.6; 40.2; 36.1; 35.4; 29.9; 28.8; 26.7; 26.4; 26.3; 25.4; 25.3; 24.9; 24.5; 22.8; 21.8. Mass spectrum (ES-API), m/z: 212 [M–HCl+H]+. Found, %: C 58.21; H 9.08; N 5.55; Cl 14.63. C12H22ClNO2. Calculated, %: C 58.17; H 8.95; N 5.65; Cl 14.31.
Potassium cis-4-(piperidin-4-yl)tetrahydro-2H-pyran-3-carboxylate (8c) was obtained as a single cis-diastereomer by crystallization. Yield 339 mg (45%), yellowish solid, mp >300°C. 1H NMR spectrum (500 MHz, D2O), δ, ppm (J, Hz): 3.99 (1H, d, J = 11.6, H-5); 3.86 (1H, d, J = 11.6, H-1); 3.46 (1H, dd, J = 11.6, J = 3.0, H-5); 3.32 (1H, td, J = 11.6, J = 2.5, H-1); 3.19–3.04 (2H, m, H-11,15); 2.71–2.57 (2H, m, H-11,15); 2.44–2.34 (1H, m, H-2); 1.93–1.74 (3H, m, H-4,12,14); 1.61–1.53 (1H, m, H-13); 1.47–1.41 (1H, m, H-4); 1.40–1.31 (1H, m, H-3); 1.16–1.00 (2H, m, H-12,14); NH signal is not visible due to the exchange with D2O. 13C NMR spectrum (151 MHz, D2O), δ, ppm: 181.6; 70.3; 68.0; 44.7; 44.6; 40.8; 36.3; 28.1; 28.0; 25.1. Mass spectrum (ES-API), m/z: 212 [M–K]–. Found, %: C 52.29; H 7.59; N 5.44. C11H18KNO3. Calculated, %: C 52.56; H 7.22; N 5.57.
Potassium 1'-(tert-butoxycarbonyl)[3,4'-bipiperidine]-4-carboxylate (9) was synthesized according to general method for the synthesis of amino acids 8a–c from compound 5d (0.500 mmol). The compound was obtained as a single trans-diastereomer by crystallization. Yield 70.1 mg (40%), yellowish solid, mp >300°C. 1H NMR spectrum (600 MHz, D2O), δ, ppm (J, Hz): 4.03–3.91 (2H, m, H-8,12); 3.32 (1H, d, J = 12.8, H-6); 3.28 (1H, dd, J = 12.8, J = 3.8, H-2); 2.84 (1H, td, J = 13.1, J = 3.3, H-6); 2.73–2.56 (3H, m, H-2,8,12); 2.37 (1H, td, J = 11.7, J = 3.7, H-4); 2.02–1.97 (1H, m, H-5); 1.87–1.82 (1H, m, H-3); 1.76–1.69 (1H, m, H-5); 1.60–1.51 (2H, m, H-10,11); 1.46–1.41 (1H, m, H-9); 1.37–1.14 (10H, m, C(CH3)3, H-9); 1.03–0.97 (1H, m, H-11); NH signal is not visible due to the exchange with D2O. 13C NMR spectrum (151 MHz, D2O), δ, ppm: 181.0; 156.4; 81.6; 45.8; 44.0; 43.2; 39.9; 37.0; 29.3; 27.6; 26.3; 26.0. Mass spectrum (ES-API), m/z: 311 [M–K]–. Found, %: C 55.04; H 7.49; N 8.28. C16H27KN2O4. Calculated, %: C 54.83; H 7.76; N 7.99.
Supplementary information file containing 1H and 13C NMR spectra of the synthesized compounds is available at the journal website http://springerlink.bibliotecabuap.elogim.com/journal/10593.
The work was funded by Enamine Ltd. An additional funding was received from Ministry of Education and Sciences of Ukraine, grants No. 0121U100387 (21BF037-01M) and 0122U001962 (22BF037-02).
The authors thank Prof. Andrey A. Tolmachev for his encouragement and support and the brave people of Ukraine who made finalizing this publication possible.
References
Han, J.; Konno, H.; Sato, T.; Soloshonok, V. A.; Izawa, K. Eur. J. Med. Chem. 2021, 220, 113448.
Schweizer, F. Angew. Chem., Int. Ed. 2002, 41, 230.
Liu, J.; Han, J.; Izawa, K.; Sato, T.; White, S.; Meanwell, N. A.; Soloshonok, V. A. Eur. J. Med. Chem. 2020, 208, 112736.
Kiss, L.; Mándity, I. M.; Fülöp, F. Amino Acids 2017, 49, 1441.
Feskov, I. O.; Golub, B. O.; Vashchenko, B. V.; Levterov, V. V.; Kondratov, I. S.; Grygorenko, O. O.; Haufe, G. Eur. J. Org. Chem. 2020, 4755.
Hys, V. Y.; Shevchuk, O. I.; Vashchenko, B. V.; Karpenko, O. V.; Gorlova, A. O.; Grygorenko, O. O. Eur. J. Org. Chem. 2020, 3896.
Barbier, P.; Renzetti, A. R.; Turbanti, L.; Di Bugno, C.; Fornai, F.; Vaglini, F.; Maggio, R.; Corsini, G. U. Eur. J. Pharmacol. Mol. Pharmacol. 1995, 290, 125.
Subissi, A.; Alberto Maggi, C.; Meli, A. Jpn. J. Pharmacol. 1986, 42, 153.
Ali, A.; Jadhav, A.; Jangid, P.; Patil, R.; Shelar, A.; Karuppayil, S. M. J. Antibiot. (Tokyo) 2018, 71, 456.
Dom, R.; Lommel, R.; Baro, F. Acta Psychiatr. Scand. 1971, 47, 399.
van Wijngaarden, I.; Soudijn, W. Life Sci. 1968, 7, 225.
van Wijngaarden, I. Life Sci. 1969, 8, 517.
DeForrest, J. M.; Waldron, T. L.; Harvey, C.; Scalese, B.; Rubin, B.; Powell, J. R.; Petrillo, E. W.; Cushman, D. W. J. Cardiovasc. Pharmacol. 1989, 14, 730.
Murdoch, D.; McTavish, D. Drugs 1992, 43, 123.
Pilote, L.; Abrahamowicz, M.; Eisenberg, M.; Humphries, K.; Behlouli, H.; Tu, J. V. Can. Med. Assoc. J. 2008, 178, 1303.
Subota, A. I.; Lutsenko, A. O.; Vashchenko, B. V.; Volochnyuk, D. M.; Levchenko, V.; Dmytriv, Y. V.; Rusanov, E. B.; Gorlova, A. O.; Ryabukhin, S. V.; Grygorenko, O. O. Eur. J. Org. Chem. 2019, 3636.
ElMarrouni, A.; Ritts, C. B.; Balsells, J. Chem. Sci. 2018, 9, 6639.
López, E.; Melis, C.; Martín, R.; Petti, A.; Hoz, A.; Díaz-Ortíz, Á.; Dobbs, A. P.; Lam, K.; Alcázar, J. Adv. Synth. Catal. 2021, 363, 4521.
Subotin, V. V.; Vashchenko, B. V.; Asaula, V. M.; Verner, E. V.; Ivanytsya, M. O.; Shvets, O.; Ostapchuk, E. N.; Grygorenko, O. O.; Ryabukhin, S. V.; Volochnyuk, D. M.; Kolotilov, S. V. Molecules 2023, 28, 1201.
Cai, C.-Y.; Wu, Z.-J.; Liu, J.-Y.; Chen, M.; Song, J.; Xu, H.-C. Nat. Commun. 2021, 12, 3745.
Mozingo, R. Org. Synth. 1946, 26, 77.
Grygorenko, O. O.; Volochnyuk, D. M.; Vashchenko, B. V. Eur. J. Org. Chem. 2021, 6478.
Armarego, W. L. F.; Chai, C. Purification of Laboratory Chemicals; Elsevier: Oxford, 2003.
Hsu, D.-S.; Liang, S.-P. J. Org. Chem. 2020, 85, 1270.
Peil, S.; Fürstner, A. Angew. Chem., Int. Ed. 2019, 58, 18476.
Su, N.; Theorell, J. A.; Wink, D. J.; Driver, T. G. Angew. Chem., Int. Ed. 2015, 54, 12942.
Whelligan, D. K.; Solanki, S.; Taylor, D.; Thomson, D. W.; Cheung, K.-M. J.; Boxall, K.; Mas-Droux, C.; Barillari, C.; Burns, S.; Grummitt, C. G.; Collins, I.; van Montfort, R. L. M.; Aherne, G. W.; Bayliss, R.; Hoelder, S. J. Med. Chem. 2010, 53, 7682.
Binanzer, M.; Hsieh, S.-Y.; Bode, J. W. J. Am. Chem. Soc. 2011, 133, 19698.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2023, 59(6/7), 442–448
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Verner, E.V., Vashchenko, B.V., Sosunovych, B. et al. Novel approach to saturated amino acid derivatives with isolated (hetero)cyclic rings via the hydrogenation of dienes. Chem Heterocycl Comp 59, 442–448 (2023). https://doi.org/10.1007/s10593-023-03214-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-023-03214-x