

The reactions of dichlorodimethylsilane or 1,3-dichlorotetramethyldisiloxane with bis(trimethylsilyl) ester of terephthalic acid (1:1 molar ratio, 20°C, 72–144 h) gave 69–97% yields of previously unknown 27- and 22-membered cyclic organosilicon esters of terephthalic acid, respectively. The molecular structures of 22-membered ester 4,4,6,6,12,12,14,14-octamethyl-3,5,7,11,13,15-hexaoxa-4,6,12,14-tetrasila-1,9(1,4)-dibenzacyclohexadecaphane-2,8,10,16-tetraone and trimethylsilyl ester of terephthalic acid were studied by X-ray structural analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Organosilicon derivatives of terephthalic acid, in contrast to organic derivatives,2,3 have not yet been thoroughly studied. It is only known that a polymeric electrolyte on the basis of boron and bis(trimethylsilyl)-terephthalate is used in rechargeable lithium ion batteries,4 while bis(trimethylsilyl)terephthalate itself acts as a corrosion inhibitor.5
We have previously shown6 that the reactions between trimethylsilyl ester of phthalic acid (1) and methyl-(chloromethyl)- or dichloromethyl(phenyl)silane at room temperature gave nearly quantitative yields of 7-membered cyclic organosilicon esters – 3-chloromethyl-3-methyl- and 3-methyl-3-phenyl-2,4,3-benzodioxasilepine-1,5-dione, respectively (Scheme 1).

Scheme 1
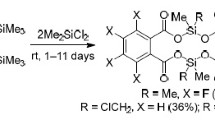
The reactions between trimethylsilyl ester of 2-(trimethylsiloxy) benzoic acid and organosilicon halides RSiX3–nYn (R = Me, CH=CH2, Ph; X, Y = Cl; X = F, Y = Cl) proceeded similarly, leading to the formation of 6-membered cyclic organosilicon esters of salicylic acid7 (Scheme 2).

Scheme 2
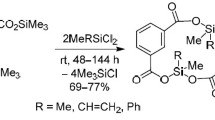
The reactions between bis(trimethylsilyl) ester of isophthalic acid and dichloromethyl(organyl)silanes MeRSiCl2 (R = Me, CH=CH2, Ph; 1:1 molar ratio, 20–25°С) proceeded differently from the reactions of ester 1 with similar dichlorosilanes and led to the formation of 16-membered heterocycles (silacyclophanones) – 4,10-dimethyl-4,10-diorganyl-3,5,9,11-tetraoxa-4,10-disila-1,7(1,3)-dibenzacyclododecaphane-2,6,8,12-tetraones1 (Scheme 3).

Scheme 3
While continuing research in this direction, we established that the reaction of dichlorodimethylsilane with bis(trimethylsilyl) ester of terephthalic acid (2) (Fig. 1) (1:1 molar ratio, 20°C, 144 h) provided a previously unknown 27-membered cyclic organosilicon ester of terephthalic acid – 4,4,10,10,16,16-hexamethyl-3,5,9,11,15,17-hexaoxa-4,10,16-trisila-1,7,13(1,4)-tribenzacyclooctadecaphane-2,6,8,12,14,18-hexaone (3) in 69% yield (Scheme 4). The formation of this product was proved by methods of NMR spectroscopy, mass spectrometry, and elemental analysis.

The molecular structure of bis(trimethylsilyl) ester of terephthalic acid (2) with atoms represented by thermal vibration ellipsoids of 50% probability.

Scheme 4
The interaction of 1,3-dichlorotetramethyldisiloxane with ester 2 (1:1 molar ratio, 20°C, 72 h) gave 97% yield of a previously unknown 22-membered cyclic organosilicon ester of terephthalic acid – 4,4,6,6,12,12,14,14-octamethyl-3,5,7,11,13,15-hexaoxa-4,6,12,14-tetrasila-1,9(1,4)-dibenzacyclohexadecaphane-2,8,10,16-tetraone (4) (Scheme 5). Its formation was proved by methods of X-ray structural analysis (Fig. 2), NMR spectroscopy, mass spectrometry, and elemental analysis. The 3.561 Å distance between the centroids of С=С bond and the unsaturated ring pointed to a π-stacking interaction between the phenyl rings.

Scheme 5

The molecular structure of ester 4 with atoms represented by thermal vibration ellipsoids of 50% probability. The red circles connected by dotted line indicate the 3.561 Å distance between the centroids of С=С bond and the unsaturated ring.
The obtained cyclic esters 3 and 4 were isolated as amorphous and crystalline materials, respectively. They were stable during storage under inert atmosphere and were readily soluble in the majority of polar organic solvents (CHCl3, Et2O, MeCN).
Thus, we have developed an effective method for the preparation of previously unknown cyclic organosilicon esters of terephthalic acid.
Experimental
IR spectra were recorded on a Specord IR 75 spectrometer in KBr pellets. 1H, 13С, and 29Si NMR spectra were acquired on a Bruker DPX 400 instrument (400, 100, and 162 MHz, respectively) in CDCl3, using TMS as internal standard. Mass spectra were recorded on a Shimadzu GCMS-QP5050A instrument, injector temperature 200–250°С, the carrier gas was helium, detector temperature 200°С, quadrupole mass analyzer, EI ionization (70 eV). Chromatographic separation of the compounds was performed on an SPB-5 capillary column (60 m × 0.25 mm × 0.25 μm), carrier gas helium, flow rate 0.7 ml/min, evaporator temperature 230°С, ion source temperature 200°С, pressure 280 kPa, temperature program from 60 to 250°С at the rate of 10°/min. Elemental analysis (C, H) was performed on a Flash EA 1112 Series elemental analyzer. The content of silicon was determined according to the procedure described by Gelman.8 Melting points were determined by using a Kofler bench.
Dichlorodimethylsilane was commercially available and was purified by distillation prior to use. 1,3-Dichlorotetramethyldisiloxane was synthesized according to a published procedure.9 Bis(trimethylsilyl) ester of terephthalic acid (2) was synthesized by a different literature procedure.6, 10 Its physical properties matched the literature data.11, 12
4,4,10,10,16,16-Hexamethyl-3,5,9,11,15,17-hexaoxa-4,10,16-trisila-1,7,13(1,4)-tribenzacyclooctadecaphane-2,6,8,12,14,18-hexaone (3). A mixture of dichlorodimethylsilane (1.29 g, 0.01 mol) with bis(trimethylsilyl) ester of terephthalic acid (2) (3.10 g, 0.01 mol) was maintained at room temperature for 144 h. The reaction was performed without solvent in a flask equipped with reflux condenser under argon atmosphere. Chlorotrimethylsilane formed during the reaction (1.93 g, 89%) was decanted from the obtained precipitate, the precipitate was washed with heptane and dried at reduced pressure. Yield 1.53 g (69%), white amorphous material. IR spectrum, ν, cm–1: 3075 (C–H C6H4), 2975, 2915 (C–H CH3), 1960, 1720, 1695 (C=O), 1595, 1450 (C=C C6H4), 1291, 1133 (Si–O), 831 (Si–CH3). 1H NMR spectrum, δ, ppm: 0.52 (18H, s, 6CH3); 7.36–7.41 (12H, m, 3C6H4). 13C NMR spectrum, δ, ppm: –1.4 (CH3); 129.9; 130.0; 130.2; 130.4; 133.9; 134.2; 134.5; 135.6 (C6H4); 165.6 (C=O). 29Si NMR spectrum, δ, ppm: 8.47. Mass spectrum, m/z (Irel, %): 666 [M]+ (0.2), 651 [M–Me]+ (16), 577 [M–Me–Me2SiO]+ (3), 517 [M–C6H4(CO)(COOH)]+ (2), 503 (11), 443 [M–C6H4(COOSiMe2)(COOH)]+ (1), 429 (28), 369 [M–C6H4(COOSiMe2)(COOH)–Me2SiO]+ (3), 295 [C6H4(COOSiMe2)(COOSiMe2H)]+ (10), 281 (4), 267 (2), 223 [C6H4(COOSiMe2)(COOH)]+ (44), 177 (11), 166 (48), 149 [C6H4(CO)(COOH)]+ (100), 133 (5), 121 [C6H4(COOH)]+ (20), 93 [C6H4OH)]+ (4), 75 [Me2SiOH]+ (16). Found, %: С 54.11; Н 4.33; Si 12.16. C30H30O12Si3. Calculated, %: С 54.04; H 4.53; Si 12.64.
4,4,6,6,12,12,14,14-Octamethyl-3,5,7,11,13,15-hexaoxa-4,6,12,14-tetrasila-1,9(1,4)-dibenzacyclohexadecaphane-2,8,10,16-tetraone (4) was synthesized analogously from ester 2 (1.29 g, 0.01 mol) and 1,3-dichlorotetramethyldisiloxane (2.03 g, 0.01 mol). The precipitated product was recrystallized from CHCl3. Yield 2.83 g (97%), colorless crystals, mp 185–186°С. IR spectrum, ν, cm–1: 3060 (C–H C6H4), 2974 (C–H CH3), 1705 (C=O), 1626, 1410 (C=C C6H4), 1263, 1069 (Si–O), 817 (Si–CH3). 1H NMR spectrum, δ, ppm: 0.47 (24H, s, 8CH3); 7.25–8.14 (8H, m, 2C6H4). 13C NMR spectrum, δ, ppm: –0.4 (CH3); 129.7; 134.5 (C6H4); 164.9 (C=O). 29Si NMR spectrum, δ, ppm: –5.2.
Mass spectrum, m/z (Irel, %): 592 [M]+ (2), 577 [M–Me]+ (100), 429 [M–Me–C6H4(CO)2O]+ (2), 327 (2), 295 (2), 281 [M–Me–2C6H4(CO)2O]+ (54), 267 (2), 253 (6), 251 (2), 236 (2), 223 (3), 214 (2), 207 (2), 193 (2), 179 (4), 166 (9), 149 [C6H4(CO)(COOH)]+ (18), 133 (9), 129 (2), 121 (5), 118 (6), 103 (31), 90 (2), 75 [Me2SiOH]+ (17). Found, %: С 48.14; Н 5.55; Si 18.52. C24H32O10Si4. Calculated, %: С 48.62; H 5.44; Si 18.95.
X-ray structural analysis of compounds 2, 4 was performed on a Bruker D8 Venture monocrystal X-ray diffractometer with a Photon 100 detector (ω–φ-scanning). The intensity of reflections was integrated using Bruker SAINT software. The X-ray absorption of the crystal was accounted for by analyzing the intensity of equivalent reflections. After averaging the intensities of equivalent reflexes, only independent reflexes were used. The search for model was performed by direct methods using the SHELXS program.13 As a result, the coordinates of all nonhydrogen atoms were determined. The hydrogen atom positions were calculated from difference Fourier maps. The obtained structure was refined by the method of least squares using the SHELXL program.13 The crystallographic datasets for compounds 2, 4 were deposited at the Cambridge Crystallographic Data Center (deposits ССDС 1553758 and ССDС 1846662, respectively).
References
Basenko, S. V.; Soldatenko, A. S. Chem. Heterocycl.Compd. 2018, 54, 100. [Khim. Geterotsikl. Soedin. 2018, 54, 100.]
Lorz, P. M.; Towae, F. K.; Enke, W.; Jäckh, R.; Bhargava, N.; Hillesheim, W. In Ullmann's Encyclopedia of Industrial Chemistry; Wiley, 2007.
Sheehan, R. J. In Ullmann's Encyclopedia of Industrial Chemistry; Wiley, 2011.
Takayuki, T.; Hiroaki, W.; Atsuki, S.; Akiko, T.; Eiji, K. WO Patent 2013065723.
Liston, T. V. US Patent 3538000.
Basenko, S. V.; Bormashev, P. A.; Mirskov, R. G.; Voronkov, M. G. Dokl. Chem. 1993, 331, 173. [Dokl. Akad. Nauk 1993, 331, 177.]
Basenko, S. V.; Zelenkov, L. E. Chem. Heterocycl.Compd. 2015, 51, 295. [Khim. Geterotsikl. Soedin. 2015, 51, 295.]
Gelman, N. E. Methods of Quantitative Organic Elemental Microanalysis [in Russian]; Khimiya: Moscow, 1987, p. 165.
Basenko, S. V.; Soldatenko, A. S.; Voronkov, M. G. Dokl. Chem. 2013, 451, 203. [Dokl. Akad. Nauk 2013, 451, 528.]
Liston, T. V. US Patent US3631084.
Henglein, F. A.; Abelsnes, G.; Heneka, H.; Lienhard, Kl.; Nakhre, Pr.; Scheinost, K. Makromol. Chem. 1957, 24, 1.
Voronkov, M. G.; Dolmaa, G.; Putilova, G. G.; Dubinskaya, E. I. Zh. Obshch. Khim. 1985, 55, 2297.
Sheldrick, G. M. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, A64, 112.
The main results of this study were obtained by using the equipment at the Collective use analytical center “Baikal”, Siberian Branch of the Russian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Khimiya Geterotsiklicheskikh Soedinenii, 2018, 54(8), 826–828
* For Communication 1, see 1.
Rights and permissions
About this article

Cite this article
Basenko, S.V., Soldatenko, A.S., Vashchenko, O.V. et al. Silacyclophanones 2*. Cyclic organosilicon esters of terephthalic acid. Chem Heterocycl Comp 54, 826–828 (2018). https://doi.org/10.1007/s10593-018-2357-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-018-2357-0