

A new eco-friendly and reliable methodology for the synthesis of 1,2,3,4-tetrahydroquinazolines is proposed. This simple protocol was tested for the continuous synthesis of a small library of 1,2,3,4-tetrahydroquinazoline derivatives demonstrating good versatility and applicability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
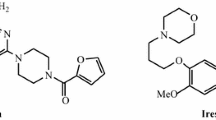
Nitrogen-containing heterocycles represent a significant structural motif widely distributed in biologically active natural and synthetic compounds.1 Among the others, quinazolines and 1,2,3,4-tetrahydroquinazolines have attracted a large interest from both the synthetic and pharmaceutical points of view. In fact, the quinazoline scaffold is a part of numerous pharmaceutically active compounds (Fig. 1), and in particular, 1,2,3,4-tetrahydro derivatives display antiinflammatory and analgesic properties.2

Biologically active quinazoline derivatives.
The quinazoline moiety is easily prepared by 1,2,3,4-tetrahydroquinazoline oxidation,3 that, in turn, can be obtained in the condensation reaction between 2-aminobenzylamine and aldehydes or ketones. This reaction has been explored by several research groups, the desired product has been obtained for the first time by refluxing the reagents in benzene or xylene with azeotropic removal of water.4 In order to improve the green aspect of the synthesis, ionic liquids have been explored as reaction medium,5 and more recently, Scott et al. proposed a protocol which involves ''water slurry'' or ''solvent-free'' conditions.6 It was also demonstrated that 2-aryl-substituted derivatives can exhibit a ring-chain tautomeric equilibrium depending on the nature of the substituent and the solvent.7 Flow chemistry in the past decades has received a growing attention because of a number of benefits over traditional batch techniques8 with interesting applications in combinatorial chemistry and library synthesis.9
As an extension of our interest in the synthesis of heterocycles,10 including flow techniques,11 we optimized the conditions for the fast preparation of a library of substituted 1,2,3,4-tetrahydroquinazoline derivatives using a flow setup.
The results of a preliminary screening of the reaction conditions, using as model the reaction between diamine 1 and p-fluorobenzaldehyde (2a) in a 2-ml tubular reactor filled with the promoter of the reaction are summarized in Figure 2 and Table 1.

Preliminary reaction condition investigations for the synthesis of compound 3a.
Considering that the cyclization proceeds as a condensation of the Schiff base intermediate (not shown) we attempted to promote the reaction by removing the water from the passage by a drying agent like sodium sulfate (entries 1–3). It was observed that, in these cases, the solvent used as carrier influenced the outcome of the reaction and the best conversion was obtained in MeOH (28%), in comparison to EtOAc (23%), whereas in THF the 1H NMR of the crude showed the presence of a complex reaction mixture. Despite the very low solubility of the reagents it was reported that the reaction can be conveniently carried out in water and that the reaction times under ''water slurry'' conditions are, in some cases and depending on the nature of the substituents on the aromatic ring of the aldehydes, similar to those observed in ''solvent-free'' conditions under gentle heating.6 Suggesting that, most probably, hydrophobic effects and/or hydrogen bonding could facilitate the reaction affording the desired product in good yield and short reaction time. Entry 4 reports the results obtained when the reactor was filled with alumina grade V, obtained by the addition of 15% w/w of water to neutral alumina. Using these conditions, diamine 1 was converted into 1,2,3,4-tetrahydroquinazoline 3a in 99% yield after a 3 min residence time and it was demonstrated that flow rate can be increased till obtaining the same conversion yield with a 0.2 min residence time. To reduce the absorption of the products on alumina a polar solvent mixture was used (MeOH–EtOAc, 4:1).
Considering that alumina and water are not consumed during the reaction and that the starting materials were not adsorbed by the column and quantitatively converted into final products we envisioned the possibility to flux sequentially freshly prepared mixture of diamine 1 with different aldehydes 2a–g and ketones 2h,i to prepare a library of 1,2,3,4-tetrahydroquinazolines 3a–i variously substituted at the C-2 atom with aromatic and aliphatic substituents. Instantaneously prepared solutions of equimolar amounts of compounds 1 and 2a–i in MeOH were injected every 6 min via a T-junction in a stream of EtOAc generated with a variable speed FMI-LAB pump and fluxed through a 2-ml tubular reactor filled with alumina grade V collecting 30 ml of solvent after each injection (Fig. 3).

General setup of the experiment for the synthesis of compounds 3a–i.
Considering that in these conditions the reaction is very fast, a flow rate of 10 ml/min (corresponding to 0.2 min residence time) was applied enabling a very rapid preparation of the desired library of compounds 3a–i.
The results obtained exploring the scope of the reaction are collected in Table 2 evidencing an excellent efficiency of the protocol with aldehydes containing both electron-withdrawing and electron-donating groups (entries 1–7). Moderate to good results were obtained using ketones (entries 8, 9), as a consequence of the reduced reactivity of the carbonyl derivative affording lower conversion yields. In comparison to the results reported in literature,6 the reaction promoted by alumina grade V in our flow conditions is considerably faster than the same reaction performed under ''water slurry'' or ''solvent-free'' conditions. This is particularly evident in substrates having strong and weak electron-withdrawing group. As an example, compounds 3c,g can be prepared under ''water slurry'' conditions with 99 and 90% conversion but in 3 and 18 h, respectively.6,7
This indicates that alumina has an active role in the catalysis of the reaction probably in both steps of the known mechanism: activating the C=O bond and removing water during the formation of the Schiff base and H Ar); 7.08 (1H, t, J = 7.5, H Ar); 6.96 (1H, d, J = 7.4, H Ar); 6.75 (1H, t, J = 7.3, H Ar); 6.61 (1H, d, J = 7.9, H Ar); 5.23 (1H, s, CH); 4.24 (1H, d, J = 16.8, CH2); 4.19 (1H, br. s, NH); 3.96 (1H, d, J = 16.7, CH2); 2.0 (1H, br. s, NH). 13C NMR spectrum (50 MHz), δ, ppm: 143.3; 140.6; 131.8; 128.4; 127.4; 126.2; 122.4; 121.3; 118.4; 115.1; 68.9; 46.1.
2-Phenyl-1,2,3,4-tetrahydroquinazoline (3d).7 Yield 42 mg (42%), pale-yellow crystals, mp 99–101°C (mp 98–101°C7). 1H NMR spectrum (400 MHz), δ, ppm (J, Hz): 7.56–7.54 (2H, m, H Ar); 7.45–7.36 (3H, m, H Ar); 7.09 (1H, t, J = 7.6, H Ar); 6.93 (1H, d, J = 7.3, H Ar); 6.75 (1H, t, J = 7.3, H Ar); 6.62 (1H, d, J = 7.9, H Ar); 5.25 (1H, s, CH); 4.30 (1H, d, J = 16.7, CH2); 4.25 (1H, br. s, NH); 4.03 (1H, d, J = 16.7, CH2); 2.0 (1H, br. s, NH). 13C NMR spectrum (101 MHz), δ, ppm: 144.2; 142.0; 129.2; 129.0; 127.7; 127.1; 126.7; 121.7; 118.6; 115.5; 70.0; 46.9.
2-(4-Methylphenyl)-1,2,3,4-tetrahydroquinazoline (3e).12 Yield 72 mg (67%), white crystals, mp 95–96°C (mp 105–107°C12). 1H NMR spectrum (200 MHz), δ, ppm (J, Hz): 7.43 (2H, d, J = 7.8, H Ar); 7.28–7.19 (2H, m, H Ar); 7.07 (1H, t, J = 7.4, H Ar); 6.96 (1H, d, J = 7.4, H Ar); 6.74 (1H, t, J = 7.4, H Ar); 6.60 (1H, d, J = 7.4, H Ar); 5.24 (1H, s, CH); 4.28 (1H, d, J = 16.0, CH2); 4.25 (1H, br. s, NH); 4.06 (1H, d, J = 16.0, CH2); 2.38 (3H, s, CH3); 2.20 (1H, br. s, NH). 13C NMR spectrum (50 MHz), δ, ppm: 144.3; 139.4; 138.5; 129.8; 127.7; 126.9; 126.6; 121.9; 118.5; 115.7; 69.9; 46.9; 21.5.
2-(4-Methoxyphenyl)-1,2,3,4-tetrahydroquinazoline (3f).7 Yield 63 mg (55%), pale-yellow crystals, mp 104–106°C (mp 105–107°C7) 1H NMR spectrum (200 MHz), δ, ppm (J, Hz): 7.46 (2H, d, J = 8.5, H Ar); 7.11–7.02 (1H, m, H Ar); 6.98–6.92 (3H, m, H Ar); 6.74 (1H, t, J = 7.3, H Ar); 6.59 (1H, d, J = 7.9, H Ar); 5.21 (1H, s, CH), 4.29 (1H, d, J = 16.6, CH2); 4.15 (1H, br. s, NH); 4.00 (1H, d, J = 16.6, CH2); 3.84 (3H, s, OCH3); 3.83 (1H, br. s, NH). 13C NMR spectrum (101 MHz), δ, ppm: 159.2; 143.3; 133.3; 127.2; 126.7; 125.7; 120.7; 117.6; 114.5; 113.5; 68.6; 54.8; 46.0.
2-(4-Nitrophenyl)-1,2,3,4-tetrahydroquinazoline (3g).7 Yield 101 mg (82%), white crystals, mp 102–105°C (mp 105–107°C7) 1H NMR spectrum (400 MHz), δ, ppm (J, Hz): 8.27 (2H, d, J = 8.5, H Ar); 7.76 (2H, d, J = 8.5, H Ar); 7.12 (1H, t, J = 7.9, H Ar); 6.97 (1H, d, J = 7.5, H Ar); 6.79 (1H, t, J = 7.4, H Ar); 6.68 (1H, d, J = 8.0, H Ar); 5.41 (1H, s, CH); 4.21 (1H, d, J = 16.6, CH2), 4.20 (1H, br. s, NH), 3.93 (1H, d, J = 16.7, CH2); 2.0 (1H, br. s, NH). 13C NMR spectrum (101 MHz), δ, ppm: 149.1; 148.3; 145.1; 128.3; 127.9; 126.7; 124.3; 121.8; 119.2; 115.8; 68.8; 45.8.
2-Methyl-2-phenyl-1,2,3,4-tetrahydroquinazoline (3h).7 The pure compound was not isolated, compound signals indicated from the spectra of the crude. 1H NMR spectrum (400 MHz), δ, ppm (J, Hz): 7.52–7.48 (5H, m, H Ar); 7.34 (1H, t, J = 7.1, H Ar); 7.14–7.06 (1H, m, H Ar); 6.74–6.65 (2H, m, H Ar); 3.79 (1H, d, J = 16.8, CH2); 3.57 (1H, d, J = 16.8, CH2); 1.65 (3H, s, CH3); 2NH signals were not detected. 13C NMR spectrum (50 MHz), δ, ppm: 145.6; 142.3; 128.4; 127.3; 127.2; 126.5; 126.1; 120.8; 117.3; 114.3; 69.7; 43.0; 32.4.
2,2-Dimethyl-1,2,3,4-tetrahydroquinazoline (3i).7 Colorless oil. The pure compound was not isolated, compound signals indicated from the spectra of the crude. 1H NMR spectrum (200 MHz), δ, ppm (J, Hz): 7.15 (1H, dd, J = 8.0, J = 7.4, H Ar); 6.95 (1H, d, J = 7.4, H Ar); 6.70 (1H, t, J = 7.4, H Ar); 6.49 (1H, d, J = 8.0, H Ar); 4.02 (2H, s, CH2); 1.40 (6H, s, CH3); 2NH signals were not detected. 13C NMR spectrum (50 MHz), δ, ppm: 142.7; 127.2; 126.1; 119.9; 117.4; 115.0; 64.5; 42.6; 28.4.
References
Dua, R.; Shrivastava, S.; Sonwane, S. K.; Srivastava, S. K. Adv. Biol. Res. 2011, 5, 120.
Okomura, K.; Oine, T.; Yamada, Y; Hayashi, G.; Nakama, M. J. Med. Chem. 1968, 11, 348.
(a) Wang, G.; Piva de Silva, G.; Wiebe, N. E.; Fehr, G. M.; Davis, R. L. RSC Adv. 2017, 7, 48848. (b) Yamaguchi, T.; Sakairi, K.; Yamaguchi, E.; Tada N.; Itoh A. RSC Adv. 2016, 6, 56892.
Kempter, G.; Ehrlichmann, W.; Plesse, M.; Lehm, U. J. Prakt. Chem. 1982, 324, 832.
Kitazume, T.; Zulfiqar, F.; Tanaka, G. Green Chem. 2000, 2, 133.
Correa, W. H.; Papadopoulos, S.; Radnidge, P.; Roberts, B. A.; Scott, J. L. Green Chem. 2002, 4, 245.
Sinkkonen, J.; Zelenin, K. N.; Potapov, A.-K. A.; Lagoda, I. V.; Alekseyev, V. V.; Pihlaja, K. Tetrahedron 2003, 59, 1939.
(a) Atodiresei, I; Vila, C.; Rueping, M. ACS Catal. 2015, 5, 1972. (b) Degennaro, L.; Carlucci, C.; De Angelis, S.; Luisi, R. J. Flow Chem. 2016, 6, 136. (c) Fanelli, F.; Parisi, G.; Degennaro, L.; Luisi, R. Beilstein J. Org. Chem. 2017, 13, 520. (d) Degennaro, L.; Maggiulli, D.; Carlucci, C.; Fanelli, F.; Romanazzi, G.; Luisi, R. Chem. Commun. 2016, 52, 9554. (e) De Angelis, S.; De Renzo, M.; Carlucci, C.; Degennaro, L.; Luisi, R. Org. Biomol. Chem. 2016, 14, 4304.
Lange, P. P.; James, K. ACS Comb. Sci. 2012, 14, 570.
(a) Palomba, M.; Vinti, E.; Marini, F.; Santi, C.; Bagnoli, L. Tetrahedron 2016, 72, 7059. (b) Palomba, M.; Rossi, L.; Sancineto, L.; Tramontano, E.; Corona, A.; Bagnoli, L.; Santi, C.; Pannecoque, C.; Tabarrini, O.; Marini, F. Org. Biomol. Chem. 2016, 14, 2015. (c) Sancineto, L.; Mangiavacchi, F.; Tidei, C.; Bagnoli, L.; Marini, F.; Scianowski, J.; Gioiello, A.; Santi, C. Asian J. Org. Chem. 2017, 6, 988.
Cerra, B.; Mangiavacchi, F.; Santi, C.; Lozza, A. M.; Gioiello, A.. React Chem. Eng. 2017, 2, 467.
Vanden Eynde, J. J.; Godin, J.; Mayence, A.; Maquestiau, A.; Anders, E. Synthesis 1993, 867.
The University of Perugia for the financial support “Fondo Ricerca di Base 2017” is warmly acknowledged. F.M. acknowledges National Consortium of the Italian University C.I.N.M.P.I.S. for the fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2018, 54(4), 478–481
Rights and permissions
About this article

Cite this article
Mangiavacchi, F., Mollari, L., Bagnoli, L. et al. Condensation of 2-aminomethylaniline with aldehydes and ketones for the fast one-pot synthesis of a library of 1,2,3,4-tetrahydroquinazolines under flow conditions. Chem Heterocycl Comp 54, 478–481 (2018). https://doi.org/10.1007/s10593-018-2292-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-018-2292-0