
Abstract
Accurate description of population structure and genetic connectivity is essential for efficient conservation efforts. Along the European coastline, Tursiops truncatus typically shows high site fidelity to relatively small areas, often semi-enclosed waters, but patterns of genetic connectivity among such areas are often poorly understood. In this study, we investigate the patterns of genetic structure and connectivity of Tursiops truncatus in the Adriatic Sea and contiguous Mediterranean, using multilocus microsatellite genotypes. We focus particularly on areas where photo-ID studies suggest the occurrence of local ‘resident communities’. Patterns of geographic structure were identified using multivariate methods, Bayesian assignment methods, and analyses of relatedness. Our results are consistent with the occurrence of communities with high site fidelity to the Gulf of Ambracia, Croatian island archipelagos, and the Gulf of Trieste. Dolphins in these regions do not fit a model of complete panmixia, but neither do they exhibit multiple discrete population units. Even for the community in the Gulf of Ambracia, which is well separated by several population genetic estimates, we can unambiguously identify individual dispersal to the most distant area in the Northern Adriatic Sea. We suggest that the population structure patterns in these animals might be best described as a stable metapopulation and discuss the implications of such a model for regional conservation efforts. The critically endangered Ambracian sub-population is particularly well differentiated, and is therefore at high risk of local extinction due to relatively small size, high degree of isolation and exposure to several anthropogenic pressures. The exact geographic boundaries of individual sub-populations cannot always be determined due to lack of sampling and low resolution of the methods used. Nevertheless, our results have important implications for effective conservation of local communities showing strong site fidelity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bottlenose dolphins (Tursiops truncatus) are well known for their complex patterns of population structure. This species is globally widespread, and often exhibits population structure across geographic distances much shorter than their dispersal ability (e.g. Natoli et al. 2005; Sellas et al. 2005; Parsons et al. 2006; Segura et al. 2006; Rosel et al. 2009; Martien et al. 2011; Lowther-Thieleking et al. 2015). This has been particularly well studied in European waters, where several genetic breaks have been described from the North Sea to the Mediterranean and Black Seas (Nichols et al. 2007; Viaud-Martinez et al. 2008; Louis et al. 2014; Gaspari et al. 2015b).
This species shows a tendency to form communities with a high degree of local site fidelity, with several such cases described in inshore semi-enclosed bodies of water, archipelagos or embayments (henceforth referred to as ‘resident communities’). Around the European continent, genetic studies have shown these to be differentiated from bottlenose dolphins inhabiting more open waters, but without any private alleles, and therefore, not genetically unique, namely in the Sado Estuary (Portugal; Fernández et al. 2011; Martinho et al. 2014), the Shannon Estuary (Ireland; Ingram and Rogan 2002; Mirimin et al. 2011) and the Moray Firth (Scotland; Parsons et al. 2002; Robinson et al. 2012). Natural populations might not always fit idealized models of either complete panmixia or full segregation into demographically independent units. Instead, they might sit somewhere along a continuum between these two models (e.g. Waples and Gaggiotti 2006) in what has been described as the “ambiguous zone” (Palsbøll et al. 2010). This appears to be the case for many such bottlenose dolphin ‘resident communities’.
Accurate identification of connectivity patterns among local resident communities is fundamental for effective conservation efforts and understanding threats. It not only enables more accurate estimates of local extinction risks (Lande 1988), but also of the likelihood that habitats will be replenished in the event of local reductions (Watson et al. 2011; Kunz et al. 2021). In the Eastern North Atlantic and in the Mediterranean Sea, bottlenose dolphin population structure has been described as a metapopulation (Nichols et al. 2007; Gaspari et al. 2015b), first proposed in the context of ‘resident communities’ documented in Ireland and around the British Isles (Ingram and Rogan 2002; Nichols et al. 2007; Robinson et al. 2012). In the Mediterranean Sea, several coastal sub-populations were found, connected by varying levels of gene flow through a wider ranging and vagrant sub-population (Gaspari et al. 2015b). Other studies also described population dynamics consistent with the metapopulation model (Louis et al. 2014; Nykänen et al. 2018),
However, details on the connectivity dynamics among putative sub-populations are lacking. This knowledge gap is more pronounced in the Mediterranean Sea, where several ‘resident communities’ are known to exist. In this context, determining the extent to which patterns of genetic structure might fit a metapopulation model is particularly relevant. Several studies have demonstrated that sexually mature individuals, which are able to disperse to other local breeding populations (colloquially called ‘floaters’), can play an important role in replenishing loss of individuals in local habitat patches (e.g. Grimm et al. 2005; Penteriani et al. 2011; Lenda et al. 2012; Robles and Ciudad 2020).
In the Adriatic Sea and contiguous Ionian Sea, several long-term photo-identification (hereafter ‘photo-ID’) studies have focused on bottlenose dolphin local ‘resident communities’. The community of the Gulf of Ambracia in Eastern Ionian Sea (Greece) shows high site fidelity and is well separated from those elsewhere in the region (Bearzi et al. 2008; Gonzalvo et al. 2016), although mid-distance dispersal out of the Gulf has been documented (Bearzi et al. 2011). Similarly, in the Gulf of Trieste and adjacent waters in the Northern Adriatic Sea, a high degree of site fidelity is shown by multiple social units (Genov et al. 2008, 2019a), which also show some level of genetic differentiation from dolphins elsewhere in the Adriatic (Gaspari et al. 2015b). The complex coastal topography of Croatian coastal regions, characterized by many islands creating archipelagos and semi-enclosed bodies of water, is also occupied by resident bottlenose dolphin communities. Photo-ID evidence suggests high site fidelity in the Kvarnerić and North Dalmatia (Northern Croatia; Bearzi et al. 1997; Pleslić et al. 2015; 2019; 2021), as well as in the Vis archipelago (Southern Croatia; Holcer 2012). Despite adjacent or even overlapping home ranges, these communities show relatively low levels of interactions (Pleslić et al. 2019). However, data on mid-distance re-sightings also indicate individual dolphins moving between different locations in the Eastern and Northern Adriatic Sea (Pleslić et al. 2015, 2019; Genov et al. 2016). These patterns are consistent with the occurrence of local populations occupying suitable habitat patches, that are visited by external ‘floaters’. However, the longer-term stability of such patterns is not fully understood, nor are the effects that these dynamics have on local and regional genetic structure.
Previous studies suggested the occurrence of genetic structure within the Adriatic Sea (Gaspari et al. 2015a), although no fine-scale estimates of recent gene flow currently exist for this region. A recent mtDNA study on bottlenose dolphins from the Gulf of Ambracia suggests a high degree of differentiation relative to other Mediterranean locations (Gonzalvo et al. 2016). Divisions between the various putative sub-populations are likely to be recent (Gaspari et al. 2015b), which makes estimating levels of differentiation and gene flow using population level methods difficult based on genetic data alone.
Consequently, identification of fine-scale gene flow dynamics in this system should rely on a more demographic level approach. Analyses of kinship and genetic relatedness within geographically restricted areas can be particularly useful in this context. Previous studies have shown the potential for kinship analyses to reveal cryptic demographic processes, particularly when combined with independent demographic data (e.g. Pilot et al. 2010; Palsbøll et al. 2010). Furthermore, if some of these bottlenose dolphin local communities have high kinship levels associated with low levels of gene-flow with other communities, they could possibly experience higher extinction risk.
In this study, we investigate the connectivity patterns of bottlenose dolphins in the Adriatic Sea and contiguous regions, by using multilocus microsatellite analyses of genetic relatedness among different potential ‘resident communities’. Our hypothesis is that putative ‘resident communities’ showing strong site-fidelity will exhibit higher levels of genetic relatedness as compared to expected averages over the broader study region. Furthermore, smaller putative ‘resident communities’ will exhibit lower genetic variation, and therefore have more uniform genetic ancestry patterns. We then discuss the observed patterns within the context of a potential metapopulation and discuss whether this model might be appropriate to use for this species in the context of local and regional conservation efforts.
Methods
Sample dataset
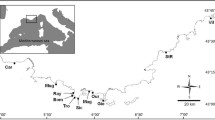
A total of 127 unique samples distributed throughout the Adriatic Sea, as well as from the Ionian and Aegean Seas, were analysed. This dataset builds upon the samples used by Gaspari et al. (2015b), by including 58.3% of novel samples not analysed before. All genotyped produced are novel, using a new genotyping protocol developed specifically for this study (see details below). Samples were divided into potential resident communities based on their geographic origin, and the pattern of population structure identified by Gaspari et al. (2015a). We considered 5 locations within the Adriatic Sea, corresponding to: (1) the Gulf of Trieste and adjacent waters (shortened as GoT elsewhere in the manuscript; Genov et al. 2008, 2019a, b) (2) the complex island archipelago in Northern Croatia, referred to as CroatiaN (Bearzi et al. 1997; Pleslić et al. 2015; 2021); (3) the Vis archipelago in Southern Croatia, referred to as CroatiaS (Holcer 2012); (4) North-Western Italian coast (from Venice to San Marino) referred to as ItalyN; (5) the Southern Italian coast of the Adriatic, (referred to as ItalyS). These five locations should also allow us to potentially capture the previously suggested East-West and North-South genetic divisions in the Adriatic (Gaspari et al. 2015b). Outside the Adriatic, samples were divided based on two well described sea basins typical of other cetacean studies in the area, namely the Ionian and Aegean Seas, and the Gulf of Ambracia as a distinct sub-basin within the Ionian Sea (Bearzi et al. 2008; Gonzalvo et al. 2016). In some analyses, three samples from the Tyrrhenian Sea were included as a comparison to the wider Mediterranean. A map showing the location of all samples and the a priori sub-population division used can be found in Fig. 1.

Map showing the distribution of samples used in this study, represented by red dots. Numbers in parenthesis reflect number of samples used in the study. Note that some samples were obtained in close proximity to one another, so number of apparent dots may be smaller than the number of actual samples
Laboratory procedures
For all samples, tissue was first cut into fine pieces using a scalpel and digested with Proteinase K at 36 °C overnight. DNA was then extracted using a GeneJet genomic DNA purification kit (Thermo Scientific), following the provided protocol. Sex of samples was determined using the protocol described in Bérubé and Palsbøll (1996).
Samples were then genotyped for a set of 16 microsatellite loci, through two multiplex PCR reactions in an ABI Verity thermocycler. Multiplex reactions were carried out using Qiagen Multiplex PCR kit with the following thermocycling conditions: 95 °C for 15 min, 40 cycles at 52 °C (primer set A)/57°C (primer set B) for 90 s, 72 °C for 1 min and a final extension at 60 °C for 30 min. Primer set A simultaneously amplified loci Dde09, Dde59, Dde65, Dde69, Dde70, Dde84 (Coughlan et al. 2006), AAT44 (Caldwell et al. 2002), Ttr58, Ttr63 (Rosel et al. 2005), while primer set B simultaneously amplified loci Dde66 (Coughlan et al. 2006), TtrRH1, TtrC12, Ttr04, Ttr11, Ttr34 (Rosel et al. 2005), KWM12a (Hoelzel et al. 1998). Successful amplification of microsatellite reaction was assessed through agarose electrophoresis stained with GelRed (Biotium). Successful amplifications were genotyped through capillary electrophoresis, using an ABI 3130 automated sequencer with the ROX500 size standard (operated by DBS Genomics at Durham University). The genotyping process was replicated for a 30% subset of all samples, to ensure reliability of the genotypes.
Data analyses
Automated sequencer trace files were analysed using the microsatellite plugin implemented in Geneious R7 (Kearse et al. 2012). Microsatellite peak calling was performed manually by two researchers independently. The dataset was checked for duplicate genotypes, and only one individual of each pair was kept for the analyses. The sample size reported earlier of 127 samples corresponds to the number of samples analysed after removing duplicates. Core genetic diversity statistics and Hardy-Weinberg equilibrium deviations tests, were calculated per locus and a priori defined populations using the software GENALEX (Peakall and Smouse 2006). The power of the microsatellite dataset to detect population structure was determined using the software PowSim (Ryman and Palm 2006). We did this by generating 1,000 simulations of populations with effective population size (Ne) values from 50 to 5,000, undergoing drift over a period of 2-100 generations. The proportion of those simulations identified as significantly differentiated using the Fisher’s Exact Test, provides a measure of the power of our microsatellite data to identify population differentiation at the average FST for each drift scenario. The power to correctly identify kinship relationships was determined using the software KinInfor (Wang 2006). We tested the following primary hypothesis/null hypothesis pairs: parent-offspring/unrelated (PO:UR), full-sibling/unrelated (FS:UR), half-sibling/unrelated (HS:UR), parent-offspring/half-sibling (PO:HS), parent-offspring/full-sibling (PO:FS) and full-sibling/half-sibling (FS:HS). Statistical significance for the power index PWR was set to 0.05, and precision to 0.01. In order to achieve a conservative estimate of power, an error rate of 0.03 for each microsatellite locus was used following the strategy used in Courbis et al. (2014).
Population structure between the a priori defined locations was assessed using two different approaches. First, we used multivariate methods without assuming a specific population genetics model. We carried out a Principal Component Analysis (PCA) using GenAlEx. An individual pairwise total genetic distance table was first created, and a PCA was created from this table using the covariance-standardized method. We also carried out a Discriminant Analysis of Principal Components (DAPC) using the R package Adegenet (Jombart 2008), to identify the most likely number of clusters. This was based on the lowest Bayesian Information Criterion value after retaining 60 principal components, and individuals were then assigned to each identified cluster based on 3 linear discriminants. Additionally, we carried out a separate DAPC assignment to all the a priori defined groups (as opposed to the 3 identified clusters) using Adegenet. This effectively acts as a multivariate individual assignment test, allowing us to investigate the distinctiveness of samples within each sub-population relative to the entire sample set.
Second, we used the software Structure (Pritchard et al. 2000) to identify the number of clusters in our dataset that maximise the Hardy-Weinberg equilibrium. We used correlated allele frequencies, and independent alphas for each cluster as recommended by Wang (2017), for cases where sampling is unequal among potential clusters. K values 1–10 were tested, through 4 independent runs of 10,000,000 iterations, with the first 25% discarded as burn-in. The most likely value of K was assessed through highest likelihood value, the ∆K method (Evanno et al. 2005), and the parsimony method (Wang 2019), all calculated using the software KFinder (Wang 2019). Convergence of results between the 4 independent runs was checked by evaluating the standard deviation of Likelihood probability for the most supported values of K, calculated using the software StructureHarvester (Earl and vonHoldt 2012).
To estimate the occurrence of close kin within and between locations, the Maximum Likelihood approach implemented in the software Colony (Jones and Wang 2010) was used to infer pairs of individuals related at the full-sibling (FS) and half-sibling (HS) level. All individuals in the dataset were considered as potential offspring, with individuals for which sex was estimated molecularly included as candidate male and female parents. To quantify whether the number of related individuals was higher than chance for a given location, we compared the number of half-sibs identified vs. the number of potential comparisons both within a location, and between a location and all other samples. Under random expectations, the number of half-sibs should increase proportionally to the number of pairwise comparisons carried out. Therefore, a linear regression line between the number of half-sibs detected and the number of pairwise comparisons available for each location should provide a description of the expected half-sibs under random expectations. If the number of half-sibs identified in a given location is above this line, then it would be higher than expected by chance.
Mean pairwise relatedness within each a priori defined location was calculated, relative to the expected mean across all samples using the Lynch and Ritland (1999) estimators (see Results of power analyses using KinInfor), as implemented in GenAlEx. This was done twice, once for all samples, and a second time after all individuals showing assignment values below 60% to the Structure clusters were removed from the dataset. This was followed by pairwise FST calculations with the software Arlequin (Excoffier et al. 2005), using the number of different alleles option, and calculation of significance with 10,100 permutations. Note that this was done in the context of a possible group of vagrant individuals that could be promoting gene flow between the various local ‘resident communities’. All admixed individuals were thus assigned to a single group designated here as “Mixed”.
The probability of origin for each individual sample was calculated jointly with estimates of gene flow between those same groupings using the software BayesAss (Wilson and Rannala 2003). Samples from stranded animals were not removed from the gene flow analyses for two key reasons: first, this would reduce the samples size of several locations to the point where they could not be used in a statistically robust manner (e.g. CroatiaN); second, because occasional or transient individuals are observed in most locations, biopsy samples may represent both high and low site-fidelity individuals (although most samples will represent photo-identified individuals with some level of site-fidelity). On the other hand, high site fidelity individuals may also strand locally. In some cases, this could be determined based on long-term photo-identification studies, but not always, and therefore it was considered more conservative to keep the samples from strandings in the analyses and consider this as a factor when interpreting gene flow estimates (numbers of biopsies per location available in Table S1). Program was run for 10,000,000 iterations following 1,000,000 of burn-in. We carried out trial runs to find mixing parameter values that could lead to mixing values for the Markov Chain Monte Carlo (MCMC) between 0.2 and 0.6, as recommended in the software manual. Convergence of the MCMC run was further assessed by analysing convergence of likelihood and ensuring all effective samples size (ESS) values were above 200, using the software Tracer (Rambaut et al. 2018).
If any of our a priori locations are part of the same genetic group, then our estimates of gene flow are effectively being calculated for sub-samples of the same population, which violates assumptions of the estimation algorithm. This can lead to biased inference if different sub-groups are an incomplete representation of the population genetic variation, due to stochastic sampling. This will be considered in our interpretation of the results obtained from this analysis, which will be done in the context of other results regarding patterns of genetic differentiation.
Results
Power simulations carried out with PowSim showed that our microsatellite dataset should be able to identify FST values of 0.01 as significant for all simulations irrespective of the Ne/t combinations. For FST values of 0.005, our microsatellites identified between 71.9% and 74.7% of simulations as significant (Table S2). Comparison with previous studies shows that our dataset achieved the statistical power expected for microsatellite-based analyses (e.g. Moura et al. 2013; Waples et al. 2018; Nykänen et al. 2018; Chabanne et al. 2021). Core genetic diversity statistics show that diversity levels are comparable among most loci and a priori locations, and there were no consistent deviations from Hardy-Weinberg across either loci or locations (Table S4). Exceptions mostly involved loci with heterozygote deficit, in locations where multiple clusters were identified in the population structure analyses (likely reflecting a Whalund effect).
Principal Component Analysis carried out in GenAlEx showed no correspondence between geographic location and clustering on the PCA plot, except for samples from Ambracia (Fig. 2A). Visually, samples from locations other than Ambracia grouped into two different clusters, one occupying the lower right quadrant of the PCA plot, and another occupying the upper half of the PCA plot. DAPC analyses identified 3 clusters as the most likely number and overlaying the distribution of the DAPC clusters on the PCA plot (Fig. 2A) shows some correspondence between the two visual PCA clusters with two of the DAPC inferred clusters, although with considerable overlap. Samples from Ambracia were all assigned to the third DAPC cluster, although it also included a few individuals from CroatiaN, CroatiaS and GoT (Fig. 2A).

PCA plot based on individual genetic distances using GENALEX. A – Samples are labelled according to location of origin. The polygons represent the distribution of the 3 clusters identified by the DAPC analyses. B – Samples are labelled according to their assignment to each of the 3 DAPC clusters. The polygons are similar to A
Both DAPC assignment and Structure showed K = 3 as the most likely number of clusters. Although K = 5 had the highest statistical support (Table 1), comparison of ancestry plots against K = 3 (best supported based on the parsimony method) showed the additional inferred clusters did not change our interpretation of population division (Fig. 3). Comparison between DAPC and Structure inference shows that only the cluster corresponding to Ambracia samples is consistent between analyses, with assignment to the other clusters disagreeing considerably. Notably, DAPC assigns most individuals to either one of the clusters, while Structure returns more admixed individuals between the two clusters (Fig. 3). DAPC assignment to the a priori locations show good separation between most locations, apart from GoT, ItalyN and Aegean, which have mixed assignment to multiple clusters, although the profiles for both GoT and ItalyN are very similar (Fig. 3).
The analyses of statistical power to detect kinship using our microsatellite dataset showed that the Lynch and Ritland (1999) estimator had the highest RMSD values (45.08) of all estimators calculated by KinInfor. Informativeness for relatedness (Ir) values ranged from 0.0268 to 0.1366 for individual loci (excluding one locus, which had an Ir of 0.005, and was therefore largely uninformative). Combined PWR values were above 0.95 for dyads PO:UR (0.9984) and FS:UR (0.9720), and above 0.6 for the dyads HS:UR (0.63), PO:HS (0.73) and FS:HS (0.64). The lowest PWR values were obtained for the most stringent dyad PO:FS (0.58). More detailed results for PWR, as well as the informativeness for relationship (IR), informativeness for relatedness (Ir) indices and respective RMSD can be found in Supplementary Table S3. These results are comparable to those reported by other microsatellite based studies (e.g. Sánchez-Montes et al. 2017; Krojerová-Prokešová et al. 2018). Although power to infer relatedness is known to be low for short tandem repeats (STR) compared to high density single nucleotide polymorphism (SNP) datasets (Kopps et al. 2015), studies have also shown that false negatives are the most likely type of error associated with this lack of power (Kozfkay et al. 2008; Kopps et al. 2015). Therefore, our estimates are conservative in terms of our interpretations regarding increased relatedness for given areas, as we are more likely to miss true relatives than to falsely identify them.

Ancestry plots obtained from DAPC and STRUCTURE analyses. Top to bottom: DAPC assignment to the a priori groups; DAPC assignment to the inferred 3 best supported clusters; Structure plot for K = 2, identified as the most likely K by the ∆K method; Structure plot for K = 3, identified as the best K by the parsimony method; Structure plot for K = 5, which had the highest likelihood value
The FullSib and HalfSib inference based on Colony showed that Ambracia has noticeably more Full/Halfsibs than any other location and considerably higher than expected by chance (Fig. 4), followed by GoT, CroatiaN, and CroatiaS. When comparing individuals across locations, CroatiaN and GoT stand out as having several pairs with other locations, although only GoT appears to have a higher number than expected by chance. GoT is also the only location that has a higher number of half-sibs both within it and with other locations. All other locations show an inverse relationship between the number of half-sibs within versus between, relative to random expectations. Most HalfSib pairings between Ambracia and other regions are with GoT, specifically with two individuals that although photo-identified and sampled in GoT, were identified by Structure as having pure Ambracian ancestry.

Top - Plot of estimated FullSib (above the diagonal) and HalfSib (below the diagonal) relationships between all samples, estimated with the software COLONY. Coloured squares show comparisons between individuals within each location. Bottom – Scatterplot of the number of half-sibs detected against the number of possible pairwise comparisons within each location and between each location and all other samples. Dark line represents the regression line for all comparisons included in the plot and represents the number of half-sibs expected by chance (see Methods for more details)
From the mean kinship analyses calculated in GenAlEx, Ambracia stands out for the clearly elevated mean kinship relative to the expected mean from all samples (Fig. 5A). Elsewhere, GoT, CroatiaN and CroatiaS have elevated values that are marginally outside the confidence limits of the test. After pooling all individuals with less than 60% assignment to the 3 clusters inferred in Structure into their own cluster (called ‘Mixed’ in the tables and figures), some locations were left with low sample size, and were therefore not included in the calculations. These locations are instead represented in the ‘Mixed’ population, which thus includes individuals from Aegean, Ionian, ItalyS and Tyrrhenian. Recalculating mean kinship for this revised sample set, it is clear that most locations now have higher than expected relatedness values (although values are relatively low), except the ‘Mixed’ cluster (Fig. 5B).

Average pairwise relatedness within each region compared to random expectations. A - all populations are included. B - all individuals with less than 60% assignment to one of the 3 clusters identified in STRUCTURE are pooled into a single population designated as Mixed. This left some locations with low sample sizes, and these are therefore not included in the calculation. Error bars around mean represent the 95% confidence interval of local estimates, calculated by bootstrapping. U and L represent the upper and lower (respectively) 95% limits of the null hypothesis of no difference in mean relatedness relative to other populations, calculated by permutation
Pairwise FST between these locations left by the revised dataset show that Ambracia is well differentiated from all other locations, with relatively high and significant FST values (all > 0.125; Table 2), and thus likely experiences stronger isolation. Within the Adriatic basin, CroatiaS has the highest FST values when compared to other Adriatic locations. All other values are at least an order of magnitude lower, with comparisons involving ItalyN, CroatiaN and GoT being particularly low (all < 0.006; Table 2).

Diagrammatic representation of patterns of gene flow between a priori locations. A- directional gene flow plot was based on the BAYESASS estimates (numerical values in Table 3). Double bars represent gene flow events that have the location as source. Plot was produced using the package CIRCLIZE (Gu et al. 2014), following the workflow presented in Sander et al. (2014), and represents only the values in the top 0.65 percentile. B- heatmap showing the probability of origin for each sample, resulting from the BAYESASS gene flow estimate
Estimates of contemporary gene flow obtained from BayesAss, are consistent with the pairwise FST values. GoT appears to be connected with other locations with relatively high levels of gene flow (22.22 − 26.33%; Table 3), while Ambracia appears to have low gene flow with other locations (1.4-1.6%; Table 3), as well as residual gene flow with GoT (2.81%; Table 3) and the ‘Mixed’ group (3.2%; Table 3). Directionality suggests that the GoT community has mostly outgoing gene flow and would therefore be mostly a source sub-population, while the cluster ‘Mixed’ shows the opposite pattern and would therefore be mostly a sink sub-population (Fig. 6A). However, these directionality estimates should be interpreted cautiously given the low genetic differentiation between locations within the Adriatic basin, and the differences in sample size among locations (GoT has the highest sample size). In this sense, estimates involving Ambracia are likely more reliable as it shows higher level of differentiation and a more balanced sample size relative to other locations. Ambracia exhibits mostly outgoing gene flow and could therefore be considered as a source sub-population (Fig. 6A), albeit with low levels of gene flow.
Analysis of the probability of origin for each individual samples help identify migration patterns more clearly. The Gulf of Ambracia, GoT and CroatiaN are the only locations which have individuals with high probability of being native, although this proportion is higher for GoT and in particular for Ambracia (where all individuals are inferred as native, and all samples were obtained through biopsies; Fig. 6B). In GoT, the two individuals with high probability of being first generation migrants from Ambracia, are the same which are identified in Structure as having Ambracian ancestry. From long term photo-ID monitoring in the area, these two individuals were seen regularly in GoT for a period of two years during 2013–2014 (but not in the preceding ten years of study). During this time, they exhibited very high site fidelity, but have subsequently not been observed in the area. Therefore, they should be considered as having low long-term site-fidelity. Individuals with some probability of being migrants in GoT were all unidentified stranded animals of unknown origin at the time of sampling. Conversely, all individuals with high probability of being native to GoT were biopsied photo-identified individuals known to occupy the area regularly. In CroatiaN, samples of individuals with high probability of being native were from both strandings and biopsies, and all individuals identified as migrants were assigned to GoT by the gene flow estimates. For ItalyN, ItalyS and CroatiaS, all individuals were identified as first-generation migrants from either GoT or CroatiaN (Fig. 6B).
We provide more details in the Discussion, but it should be noted here that these results suggest the existence of a wider-ranging Adriatic population, which is being represented (at least in part) in these analyses by both GoT and CroatiaN samples. The individuals pooled into the ‘Mixed’ group are all identified as migrants from either CroatiaN, GoT or Ambracia (Fig. 6B), which likely reflects the higher genetic diversity resulting from mixing between different locations.
Discussion
Previous studies suggested the presence of fine-scale population structure of bottlenose dolphins in the Adriatic Sea (namely, some degree of differentiation between North-South and East-West; Gaspari et al. 2015a). However, details on connectivity patterns and gene flow dynamics were incomplete, due to limited sampling and lower resolution (Gaspari et al. 2015b). In this study, our kinship-based approach targeted at areas where the occurrence of philopatric ‘resident communities’ is either well described or suspected (Bearzi et al. 1997, 2008; Genov et al. 2008, 2019a, b; Holcer 2012; Pleslić et al. 2015, 2019, 2020; Gonzalvo et al. 2016; Rako-Gospić et al. 2017), allowed these patterns to be revealed in more detail.
Regions of complex coastal topography with high degrees of enclosure of their waters are more likely to show an increased number of close kin and reduced gene flow with other locations. However, the exact patterns and rates of gene flow varied among locations. The Gulf of Ambracia shows the strongest level of differentiation, which is consistent with it being almost entirely enclosed. However, our findings suggested that it is not completely isolated, as indicated by the recent gene flow detected through different analyses. The samples obtained in GoT, CroatiaN and CroatiaS, which are all more open systems, showed higher interconnectivity in gene flow estimates, and did not readily separate through methods based on classical population genetic models. However, they all had a higher level of kinship relative to the overall population, suggesting a higher level of local site fidelity over successive generations. GoT and CroatiaN in particular, show several individuals having high probability of being native to that area as compared to other Adriatic locations. As expected, samples from locations with least complex coastline profiles, and therefore, with more open waters (such as ItalyN, ItalyS, and Ionian) showed less evidence for differentiation, and mostly contained individuals with more diverse genetic ancestry. When these were pooled together in a single grouping (named ‘Mixed’ in the corresponding Tables and Figures), they also showed low kinship levels and high probability of being first generation migrants from other coastal locations.
These results are largely consistent with local field studies on individual site fidelity. Dolphins using local areas over several years have been identified in the Gulf of Trieste and adjacent waters, CroatiaN (Kvarnerić and North Dalmatia), CroatiaS (Vis Island archipelago) and the Gulf of Ambracia (Bearzi et al. 1997, 2008; Genov et al. 2008, 2019a, b; Holcer 2012; Pleslić et al. 2015, 2019, 2021; Gonzalvo et al. 2016). On the other hand, previous studies also found the occasional visitors in CroatiaN (Pleslić et al. 2015, 2019), and with particular frequency in the GoT (Genov et al. 2008, 2019a). Within the Gulf of Ambracia there are no records of occasional visitors. However, there is evidence of individuals first seen in the Gulf of Ambracia (and therefore inferred to be native) being observed elsewhere in the region, without subsequent re-sightings within the Gulf of Ambracia (Bearzi et al. 2011; Gonzalvo et al. 2016).
Gene flow estimates also suggest the occurrence of asymmetric gene flow, consistent with results from local field ecology studies. A recent long-term mark-recapture study in the GoT found evidence of low estimated apparent survival probability in this area (Genov 2021). Apparent survival is a product of true survival but also patterns of migration. Given that the estimated survival probability for this populations is markedly lower than is typical for this species (Genov 2021), it is likely that this partly reflects permanent movement of animals out of the area. This is consistent with the genetic results presented here, suggesting GoT gene flow balance to be mostly towards other locations. However, several elements suggest that gene flow could be mediated by a sub-population that has not been accurately represented by our sampling. This includes the fact that the main cluster receiving gene flow from other locations (designated as ‘Mixed’ in the results) is composed of samples obtained in multiple locations, with individuals identified as first-generation migrants in this ‘Mixed’ group being assigned to either GoT or CroatiaN .
We should note that an important difficulty in interpreting our results is that we cannot fully identify the geographic extent nor the exact patterns of gene flow of this putative ‘Mixed’ sub-population with the communities sampled in our study. Basin-wide aerial surveys (Fortuna et al. 2018) and on-board fishing boat monitoring schemes (Fortuna et al. 2010) identified open waters of central Northern Adriatic as an area of high bottlenose dolphin abundance. Additionally, a recent study in CroatiaN and CroatiaS regions found limited interactions between three coastal communities, but a notable number of occasional visitors coming from the open part of the Adriatic Sea (Pleslić et al. 2019).
In this context, open waters of the Northern-Central Adriatic Sea may host its own sub-population connected to local communities, whose local characteristics may be shaped by cultural (Genov et al. 2019a) or geographical (Pleslić et al. 2019) features. Our results may reflect higher connectivity between GoT and this hypothetical wider ranging sub-population, especially considering that some individuals sampled in GoT were occasional or rare visitors. Furthermore, many individuals photo-identified alongshore in the GoT are also regularly sighted far offshore in the Northern Adriatic (T. Genov, pers. obs.). It should be noted that this higher connectivity could reflect higher mating, or a higher proportion of individuals from the wider sub-population being present in our sample set.
Possible metapopulation model in Adriatic bottlenose dolphins
Studies on cetaceans from other parts of the world have suggested metapopulation dynamics, namely: Tursiops spp. in Australia (Pratt et al. 2018; Manlik et al. 2019); spinner dolphins (Stenella longirostris) in the French Polynesia (Oremus et al. 2007); snubfin (Orcaella heinsohni) and humpback dolphins (Sousa sp.) in Australia (Brown et al. 2014); Atlantic spotted dolphins (Stenella frontalis) in the Atlantic Ocean (Méndez-Fernandez et al. 2018); pilot whales (Globicephala macrorhynchus) in Madeira (Alves et al. 2018); narwhals (Monodon monoceros) in Canada/Greenland (Heide-Jørgensen et al. 2013). In species where the term metapopulation has not been explicitly used, there is evidence for sub-populations connected by residual levels of gene flow to other sub-populations, which are at risk of local extinction (e.g. southern resident killer whales, Orcinus orca, Wasser et al. 2017). Bottlenose dolphins from the Shannon Estuary (Ireland) also show consistent photo-ID evidence of individuals showing high site fidelity to the Estuary and transient animals that visit only occasionally (Ludwig et al. 2021), which results in local genetic differentiation.
Although the exact definition of the term ‘metapopulation’ has changed since it was first introduced, when applied to the marine environment, it is often defined as: discrete local breeding sub-populations connected by varying levels of gene flow, and where at least one sub-population has the potential to experience extinction from stochastic events (Smedbol et al. 2002). Our results are consistent with the occurrence of multiple sub-populations, with asymmetric gene-flow patterns consistent with source-sink dynamics (Pulliam 1988), which are characteristic of metapopulations. However, a common reservation when applying a metapopulation model to marine populations relate to the difficulty in demonstrating the occurrence of repeated extinction-recolonization events (Smedbol et al. 2002; Grimm et al. 2003). This is particularly relevant to marine mammals, where long generations times (Carroll et al. 2020) make the occurrence of extinction-recolonization dynamics particularly difficult to observe or demonstrate. In cetaceans, there is precedence of fast reductions in dolphin abundance due to declining habitat (e.g. common dolphin; Piroddi et al. 2011; Moura et al. 2013) and large-scale mortality caused by culling campaigns, bycatch and epizootic events (Aguilar and Raga 1993; Bearzi et al. 2004, 2009). Studies in the British Isles showed that extant populations of bottlenose dolphin are genetically distinct from those found in Neolithic times (Nichols et al. 2007), suggesting that local extinctions of sub-populations have occurred before.
In our study area, there is some indication of fast reduction in dolphin abundance and subsequent recovery for one of the Adriatic putative sub-populations. Historically, the coastal population of bottlenose dolphins along the North-Eastern Adriatic Croatian coast have been subject to an intensive culling campaign in 1950s, when 788 animals had been killed in only five years (Bearzi et al. 2004). Although it has been proposed that most animals killed were common dolphins (D. delphis), species identification was often impossible, and the actual species ratio between the two common species (bottlenose vs. common dolphins) is unknown. Furthermore, the reported number of killed animals refers only to those landed, and it is likely that many killed or injured animals remained unreported. The impact of this source of mortality on the local resident bottlenose dolphin communities would have likely created noticeable reductions in local abundance. Recently, the Kvarnerić community, which is part of the CroatiaN sample (Fortuna 2006), showed an apparent decline of 39% between 1995 and 2003, when the local reduction risk within three generations was estimated to be 35% (Fortuna 2006). In following years (2004–2011), the same community increased in numbers and currently appears stable (although this was in part mediated by known individuals returning to the area; Pleslić et al. 2015). Although such data does not strictly represent extinction-recolonization events, the occurrence of such rapid growth cycle after local population reductions could credibly result from a metapopulation type dynamic.
Another important characteristic of metapopulations is the occurrence of ‘floaters’, sexually mature individuals that disperse into other local breeding populations. Our study confirms the occurrence of such individuals for the GoT at least, where two individuals (a male and a female) identified as having pure Ambracia ancestry, were only seen in the GoT for a limited period of time. The occurrence of such ‘floaters’ in the region is also supported by recent records of long-distance movements of one individual photo-identified in the GoT, which was also previously observed in the Tyrrhenian Sea and subsequently in the Ligurian Sea (Genov et al. 2022). Conversely, all individuals that were biopsied and known to exhibit high long-term site-fidelity, were identified as native to the area. The available data, from both genetics and photo-ID, are thus consistent with a model where inshore and more structured habitats, such as semi-enclosed bays or archipelagos, encourage the long-term settlement of individuals with high site fidelity that drive genetic differentiation, with gene flow among such locations being mediated by transient individuals that are more vagrant and make more use of open habitats. We therefore suggest that a metapopulation model best explains the available data on population connectivity and dispersal for the Adriatic and contiguous regions.
Therefore, the term metapopulation appears to be particularly useful in describing the pattern of population structure seen in Adriatic bottlenose dolphins. It is not only effective in describing population structure that is intermediate between full panmixia and complete differentiation between isolated populations, but it could also be useful in informing local and regional conservation decisions.
Conservation implications
Understanding gene flow dynamics is crucial for effective conservation management, and extinction mitigation (e.g. Carroll et al. 2020; Paquet et al. 2020; Kunz et al. 2021). Although the Gulf of Ambracia, CroatiaN, CroatiaS and the GoT appear to have different sub-population dynamics, they are all subject to anthropogenic threats to their long-term survival. This combination of high anthropogenic pressures and higher rates of emigration implies that putative sub-populations inhabiting less favourable habitats could have a relatively high local extinction risk.
Dolphins from the GoT exhibit high levels of polychlorinated biphenyls (PCBs), with most animals exceeding known toxicity thresholds and demonstrating offloading of pollutants from mothers to offspring (Genov et al. 2019b). Interactions with fisheries, both trawlers and bottom-set nets, are common and occasionally result in bycatch, while disturbance from recreational boat traffic also presents a threat (Genov et al. 2008, 2016, 2019a). The Gulf also receives substantial heavy metal discharges via the Soča/Isonzo river (Faganeli et al. 2003). Long term monitoring studies show temporally variable rates of nutrient discharge from the Soča/Isonzo river (Mozetič et al. 1998), therefore prey availability for dolphins might be quite variable in space and time as well (Genov et al. 2019a).
Threats faced by north-eastern Adriatic sub-populations are various. The Kvarnerić area, which is a Natura 2000 site for bottlenose dolphins, is subject to high seasonal acoustic pollution by nautical tourism. This causes significant displacement (Rako et al. 2013), changes in vocalisations (Rako-Gospić and Picciulin 2016) and changes in home range (Rako-Gospić et al. 2017). PCB concentrations in bottlenose dolphins from this region are among the highest found in the Adriatic and toxicological assessments indicate high health risk (Romanić et al. 2014). Coastal areas of Vis-Lastovo archipelago (CroatiaS), also a bottlenose dolphins Natura 2000 site, might have been subject to less anthropogenic pressure until recently, and contrary to areas in the Northern Adriatic, biomarker analyses suggest lower toxicological stress within the Central Adriatic area (Maltese et al. 2010; Holcer 2012). However, dolphins from this region commonly occupy areas with high trawling activity and are therefore at increased risk of bycatch and lack of prey due to overfishing (Holcer 2012).
Likewise, in the Gulf of Ambracia, dolphins are exposed to high levels of pollution (mostly derived from local agriculture) and habitat degradation (Gonzalvo et al. 2016). This Gulf is also a meso-to- eutrophic water basin, due to high nutrient discharge from the Louros and Arachthos rivers, combined with high evaporation and low water exchange with the adjacent Ionian Sea (Kountoura and Zacharias 2013). The strong differentiation and low levels of recent gene flow with other regions suggest that this community has been resident in the area for a relatively long time, and that it might be close to carrying capacity. The narrow opening to the adjacent Ionian Sea, together with a more limited prey availability due to overfishing outside of the Gulf (Gonzalvo et al. 2011, 2015), might limit dispersal into the region. Moreover, our estimates of gene flow and previous photo-identification studies (Bearzi et al. 2011; Gonzalvo et al. 2016) show that dispersal out of the Gulf is possible. Therefore, Ambracian dolphins are particularly vulnerable to inbreeding depression, as suggested by our findings of elevated kinship levels together with limited gene flow from outside locations and are therefore very likely facing a high risk of local extinction. This is consistent with the recent listing of the Gulf of Ambracia bottlenose dolphin subpopulation as Critically Endangered by IUCN Red List of Threatened Species (Gonzalvo and Notarbartolo di Sciara 2021). Recolonisation of this area by individual animals from nearby regions is unlikely, particularly if habitat quality deteriorates further.
A better understanding of connectivity, gene flow patterns and potential meta-population dynamics, such as those presented here, can help place these threats and their specific effects on different resident communities into a proper conservation context and thus inform future conservation strategies. Theoretical models suggest that under a metapopulation model, the rate of local habitat occupancy by individual floaters is dependent of local habitat quality (Kokko and Sutherland 2015). A recent study using demographic data from a passerine bird also suggested that predictions of population viability are greatly improved by taking habitat quality into account in a source-sink system (Paquet et al. 2020). This focus on habitat quality is also in line with recent calls for a broader perspective when considering cetacean conservation policies in the Mediterranean, particularly the Adriatic sea (Bearzi and Reeves 2021).
Conclusions
We provide a detailed description of population connectivity for bottlenose dolphin in the North-Eastern Mediterranean Sea. The results show that dolphins inhabiting the Gulf of Trieste, Croatian coastal areas, and the semi-enclosed waters of the Gulf of Ambracia, all contain resident communities, which are connected to dolphins elsewhere with varying levels of gene flow. Our study also implies the occurrence of a sub-population occupying wide areas of more open water habitats with connectivity to the more coastal ‘resident communities’, although its exact geographic distribution remains undetermined. We suggest that bottlenose dolphins in this region likely exhibit a stable metapopulation structure, as previously described in studies elsewhere. The Gulfs of Trieste and Ambracia are likely source sub-populations, in the sense that outgoing gene flow is higher than incoming gene flow, although estimated levels vary between the areas. The Gulf of Ambracia sub-population is particularly well differentiated, shows an elevated number of related individuals and is thus at particular risk of local extinction from increasingly intense anthropogenic pressures and potential inbreeding depression. Although restricted to the North-Eastern Mediterranean Sea, our results are similar to patterns found in other non-Mediterranean regions. A metapopulation model has important implications for the effective conservation of this species. It highlights the importance of the local suitable habitat availability, to promote and maintain viable local sub-populations. This could be particularly relevant for a species that is globally abundant, widespread, and behaviourally plastic. Although our scope was not to make recommendations for individual cases, the knowledge provided by this study will be invaluable in informing much needed local, as well as regional conservation management plans.
Code Availability
Not applicable.
References
Aguilar A, Raga JA (1993) The striped dolphin epizootic in the Mediterranean Sea. Ambio 22:524–528
Alves F, Alessandrini A, Servidio A et al (2018) Complex biogeographical patterns support an ecological connectivity network of a large marine predator in the north-east Atlantic. Divers Distrib 25:269–284. https://doi.org/10.1111/ddi.12848
Bearzi G, Reeves RR (2021) Shifting baselines of cetacean conservation in Europe. ICES J Mar Sci 78:2337–2341. https://doi.org/10.1093/ICESJMS/FSAB137
Bearzi G, Notarbartolo-DI-Sciara G, Politi E (1997) Social ecology of bottlenose dolphins in the Kvarnerić (Northern Adriatic Sea). Mar Mammal Sci 13:650–668. https://doi.org/10.1111/j.1748-7692.1997.tb00089.x
Bearzi G, Holcer D, Notarbartolo-di-Sciara G (2004) The role of historical dolphin takes and habitat degradation in shaping the present status of northern Adriatic cetaceans. Aquat Conserv Mar Freshw Ecosyst 14:363–379. https://doi.org/10.1002/aqc.626
Bearzi G, Agazzi S, Bonizzoni S et al (2008) Dolphins in a bottle: abundance, residency patterns and conservation of bottlenose dolphins Tursiops truncatus in the semi-closed eutrophic Amvrakikos Gulf, Greece. Aquat Conserv Mar Freshw Ecosyst 18:130–146. https://doi.org/10.1002/aqc.843
Bearzi G, Fortuna CM, Reeves RR (2009) Ecology and conservation of common bottlenose dolphins Tursiops truncatus in the Mediterranean Sea. Mamm Rev 39:92–123. https://doi.org/10.1111/j.1365-2907.2008.00133.x
Bearzi G, Bonizzoni S, Gonzalvo J (2011) Mid-distance movements of common bottlenose dolphins in the coastal waters of Greece. J Ethol 29:369–374. https://doi.org/10.1007/s10164-010-0245-x
Bérubé M, Palsbøll P (1996) Identification of sex in Cetaceans by multiplexing with three ZFX and ZFY specific primers. Mol Ecol 5:283–287. https://doi.org/10.1046/j.1365-294X.1996.00072.x
Brown AM, Kopps AM, Allen SJ et al (2014) Population differentiation and hybridisation of australian snubfin (Orcaella heinsohni) and Indo-Pacific humpback (Sousa chinensis) dolphins in North-Western Australia. PLoS ONE 9:e101427. https://doi.org/10.1371/journal.pone.0101427
Caldwell M, Gaines MS, Hughes CR (2002) Eight polymorphic microsatellite loci for bottlenose dolphin and other cetacean species. Mol Ecol Notes 2:393–395. https://doi.org/10.1046/j.1471-8286.2002.00270.x
Carroll EL, Hall A, Olsen MT et al (2020) Perturbation drives changing metapopulation dynamics in a top marine predator. Proc R Soc B Biol Sci 287:20200318. https://doi.org/10.1098/rspb.2020.0318
Chabanne DBH, Allen SJ, Sherwin WB et al (2021) Inconsistency between socio-spatial and genetic structure in a coastal dolphin population. Front Mar Sci 7. https://doi.org/10.3389/fmars.2020.617540
Coughlan J, Mirimin L, Dillane E et al (2006) Isolation and characterization of novel microsatellite loci for the short-beaked common dolphin (Delphinus delphis) and cross-amplification in other cetacean species. Mol Ecol Notes 6:490–492. https://doi.org/10.1111/j.1471-8286.2006.01284.x
Courbis S, Baird RW, Cipriano F, Duffield D (2014) Multiple populations of Pantropical spotted dolphins in hawaiian waters. J Hered 105:627–641. https://doi.org/10.1093/jhered/esu046
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50. https://doi.org/10.1177/11769343050010000
Faganeli J, Horvat M, Covelli S et al (2003) Mercury and methylmercury in the Gulf of Trieste (northern Adriatic Sea). Sci Total Environ 304:315–326. https://doi.org/10.1016/S0048-9697(02)00578-8
Fernández R, Santos MB, Pierce GJ et al (2011) Fine-scale genetic structure of bottlenose dolphins, Tursiops truncatus, in Atlantic coastal waters of the Iberian Peninsula. Hydrobiologia 670:111–125. https://doi.org/10.1007/s10750-011-0669-5
Fortuna CM (2006) Ecology and conservation of bottlenose dolphins (Tursiops truncatus) in the North-Eastern Adriatic Sea. PhD thesis submitted to the University of St. Andrews
Fortuna CM, Vallini C, Filidei E et al (2010) By-catch of cetaceans and other species of conservation concern during pair trawl fishing operations in the Adriatic Sea (Italy). Chem Ecol 26:65–76. https://doi.org/10.1080/02757541003627662
Fortuna CM, Cañadas A, Holcer D et al (2018) The coherence of the European Union marine natura 2000 network for wide-ranging charismatic species: a Mediterranean case study. Front Mar Sci 5:356. https://doi.org/10.3389/fmars.2018.00356
Gaspari S, Holcer D, Mackelworth P et al (2015a) Population genetic structure of common bottlenose dolphins (Tursiops truncatus) in the Adriatic Sea and contiguous regions: implications for international conservation. Aquat Conserv Mar Freshw Ecosyst 25:212–222. https://doi.org/10.1002/aqc.2415
Gaspari S, Scheinin A, Holcer D et al (2015b) Drivers of population structure of the bottlenose dolphin (Tursiops truncatus) in the Eastern Mediterranean Sea. Evol Biol 42:177–190. https://doi.org/10.1007/s11692-015-9309-8
Genov T (2021) Population ecology, behaviour and conservation status of common bottlenose dolphins (Tursiops truncatus) in the Gulf of Trieste and adjacent waters of the northern Adriatic Sea. PhD thesis submitted to the University of St. Andrews
Genov T, Kotnjek P, Lesjak J et al (2008) Bottlenose dolphins (Tursiops truncatus) in slovenian and adjacent waters (northern Adriatic Sea). Ann Ser Hist Nat 18:227–241
Genov T, Angelini V, Hace A et al (2016) Mid-distance re-sighting of a common bottlenose dolphin in the northern Adriatic Sea: insight into regional movement patterns. J Mar Biol Assoc United Kingdom 96:909–914. https://doi.org/10.1017/s0025315415001241
Genov T, Centrih T, Kotnjek P, Hace A (2019a) Behavioural and temporal partitioning of dolphin social groups in the northern Adriatic Sea. Mar Biol 166:11. https://doi.org/10.1007/s00227-018-3450-8
Genov T, Jepson PD, Barber JL et al (2019b) Linking organochlorine contaminants with demographic parameters in free-ranging common bottlenose dolphins from the northern Adriatic Sea. Sci Total Environ 657:200–212. https://doi.org/10.1016/J.SCITOTENV.2018.12.025
Genov T, Železnik J, Bruno C et al (2022) The longest recorded movement of an inshore common bottlenose dolphin (Tursiops truncatus). Mamm Biol 102:1469–1481. https://doi.org/10.1007/S42991-022-00316-5
Gonzalvo J, Notarbartolo-di-Sciara G (2021) Tursiops truncatus (Gulf of Ambracia subpopulation). In: IUCN Red List Threat. Species 2021. https://doi.org/10.2305/IUCN.UK.2021-3.RLTS.T181208820A181210985.en. Accessed 12 Nov 2022
Gonzalvo J, Moutopoulos DK, Bearzi G, Stergiou KI (2011) Fisheries mismanagement in a Natura 2000 area in western Greece. Fish Manag Ecol 18:25–38. https://doi.org/10.1111/j.1365-2400.2010.00764.x
Gonzalvo J, Giovos I, Moutopoulos DK (2015) Fishermen’s perception on the sustainability of small-scale fisheries and dolphin-fisheries interactions in two increasingly fragile coastal ecosystems in western Greece. Aquat Conserv Mar Freshw Ecosyst 25:91–106. https://doi.org/10.1002/aqc.2444
Gonzalvo J, Lauriano G, Hammond PS et al (2016) The Gulf of Ambracia’s common bottlenose dolphins, Tursiops truncatus: a highly dense and yet threatened population. Adv Mar Biol 75:259–296. https://doi.org/10.1016/BS.AMB.2016.07.002
Gospić NR, Picciulin M (2016) Changes in whistle structure of resident bottlenose dolphins in relation to underwater noise and boat traffic. Mar Pollut Bull 105:193–198. https://doi.org/10.1016/j.marpolbul.2016.02.030
Grimm V, Reise K, Strasser M (2003) Marine metapopulations: a useful concept? Helgol Mar Res 56:222–228. https://doi.org/10.1007/s10152-002-0121-3
Grimm V, Revilla E, Groeneveld J et al (2005) Importance of buffer mechanisms for population viability analysis. Conserv Biol 19:578–580. https://doi.org/10.1111/J.1523-1739.2005.000163.X
Gu Z, Gu L, Eils R et al (2014) Circlize implements and enhances circular visualization in R. Bioinformatics 30:2811–2812. https://doi.org/10.1093/bioinformatics/btu393
Heide-Jørgensen MP, Richard PR, Dietz R, Laidre KL (2013) A metapopulation model for canadian and west Greenland narwhals. Anim Conserv 16:331–343. https://doi.org/10.1111/acv.12000
Hoelzel AR, Dahlheim M, Stern SJ (1998) Low genetic variation among killer whales (Orcinus orca) in the Eastern North Pacific and genetic differentiation between foraging specialists. J Hered 89:121–128. https://doi.org/10.1093/jhered/89.2.121
Holcer D (2012) Ecology of the common bottlenose dolphin, Tursiops truncatus (Montagu, 1821) in the Central Adriatic Sea. PhD thesis submitted to the University of Zagreb
Ingram SN, Rogan E (2002) Identifying critical areas and habitat preferences of bottlenose dolphins Tursiops truncatus. Mar Ecol Prog Ser 244:247–255. https://doi.org/10.3354/meps244247
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10:551–555. https://doi.org/10.1111/j.1755-0998.2009.02787.x
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kokko H, Sutherland WJ (2015) Optimal floating and queuing strategies: consequences for density dependence and habitat loss. Am Nat 152:354–366. https://doi.org/10.1086/286174
Kopps AM, Kang J, Sherwin WB, Palsbøll PJ (2015) How well do molecular and pedigree relatedness correspond, in populations with diverse mating systems, and various types and quantities of molecular and demographic data? G3 genes, genomes. Genet 5:1815–1826. https://doi.org/10.1534/G3.115.019323
Kountoura K, Zacharias I (2013) Trophic state and oceanographic conditions of Amvrakikos Gulf: evaluation and monitoring. Desalin Water Treat 51:2934–2944. https://doi.org/10.1080/19443994.2012.748442
Kozfkay CC, Campbell MR, Heindel JA et al (2008) A genetic evaluation of relatedness for broodstock management of captive, endangered Snake River sockeye salmon, Oncorhynchus nerka. Conserv Genet 9:1421–1430. https://doi.org/10.1007/S10592-007-9466-0
Krojerová-Prokešová J, Turbaková B, Jelenčič M et al (2018) Genetic constraints of population expansion of the Carpathian lynx at the western edge of its native distribution range in Central Europe. Hered 2018 1226 122:785–799. https://doi.org/10.1038/s41437-018-0167-x
Kunz F, Kohnen A, Nopp-Mayr U, Coppes J (2021) Past, present, future: tracking and simulating genetic differentiation over time in a closed metapopulation system. Conserv Genet 22:355–368. https://doi.org/10.1007/s10592-021-01342-5
Lande R (1988) Genetics and demography in biological conservation. Science 241:1455–1460. https://doi.org/10.1126/SCIENCE.3420403
Lenda M, Maciusik B, Skórka P (2012) The evolutionary, ecological and behavioural consequences of the presence of floaters in bird populations. North West J Zool 8:394–408
Louis M, Viricel A, Lucas T et al (2014) Habitat-driven population structure of bottlenose dolphins, Tursiops truncatus, in the North-East Atlantic. Mol Ecol 23:857–874. https://doi.org/10.1111/mec.12653
Lowther-Thieleking JL, Archer FI, Lang AR, Weller DW (2015) Genetic differentiation among coastal and offshore common bottlenose dolphins, Tursiops truncatus, in the eastern North Pacific Ocean. Mar Mammal Sci 31:1–20. https://doi.org/10.1111/mms.12135
Ludwig KE, Daly M, Levesque S, Berrow SD (2021) Survival rates and capture heterogeneity of bottlenose dolphins (Tursiops truncatus) in the Shannon Estuary, Ireland. Front Mar Sci 8:301. https://doi.org/10.3389/fmars.2021.611219
Lynch M, Ritland K (1999) Estimation of pairwise relatedness with molecular markers. Genetics 152:1753–1766. https://doi.org/10.1046/j.1365-294X.2003.01835.x
Maltese S, Marsili L, Bartolini M et al (2010) Ecotoxicological status of Tursiops truncatus in the Mediterranean Sea and in the Gulf of California (Sea of Cortez-Mexico) using skin biopsy as diagnostic tool. Comp Biochem Physiol Part A Mol Integr Physiol 157:S26. https://doi.org/10.1016/j.cbpa.2010.06.075
Manlik O, Chabanne D, Daniel C et al (2019) Demography and genetics suggest reversal of dolphin source-sink dynamics, with implications for conservation. Mar Mammal Sci 35:732–759. https://doi.org/10.1111/mms.12555
Martien KK, Baird RW, Hedrick NM et al (2011) Population structure of island-associated dolphins: evidence from mitochondrial and microsatellite markers for common bottlenose dolphins (Tursiops truncatus) around the main Hawaiian Islands. Mar Mammal Sci 28:E208–E232. https://doi.org/10.1111/j.1748-7692.2011.00506.x
Martinho F, Pereira A, Brito C et al (2014) Structure and abundance of bottlenose dolphins (Tursiops truncatus) in coastal Setúbal Bay, Portugal. Mar Biol Res 11:144–156. https://doi.org/10.1080/17451000.2014.894244
Méndez-Fernandez P, Taniguchi S, Santos MCO et al (2018) Contamination status by persistent organic pollutants of the Atlantic spotted dolphin (Stenella frontalis) at the metapopulation level. Environ Pollut 236:785–794. https://doi.org/10.1016/j.envpol.2018.02.009
Mirimin L, Miller R, Dillane E et al (2011) Fine-scale population genetic structuring of bottlenose dolphins in irish coastal waters. Anim Conserv 14:342–353. https://doi.org/10.1111/j.1469-1795.2010.00432.x
Moura AE, Natoli A, Rogan E, Hoelzel AR (2013) Atypical panmixia in a european dolphin species (Delphinus delphis): implications for the evolution of diversity across oceanic boundaries. J Evol Biol 26:63–75. https://doi.org/10.1111/jeb.12032
Mozetic P, Umani SF, Cataletto B, Malej A (1998) Seasonal and inter-annual plankton variability in the Gulf of Trieste (northern Adriatic). ICES J Mar Sci 55:711–722. https://doi.org/10.1006/jmsc.1998.0396
Natoli A, Birkun A, Aguilar A et al (2005) Habitat structure and the dispersal of male and female bottlenose dolphins (Tursiops truncatus). Proc R Soc London B Biol Sci 272:1217–1226. https://doi.org/10.1098/rspb.2005.3076
Nichols C, Herman J, Gaggiotti OE et al (2007) Genetic isolation of a now extinct population of bottlenose dolphins (Tursiops truncatus). Proc R Soc London B Biol Sci 274:1611–1616. https://doi.org/10.1098/rspb.2007.0176
Nykänen M, Dillane E, Englund A et al (2018) Quantifying dispersal between marine protected areas by a highly mobile species, the bottlenose dolphin, Tursiops truncatus. Ecol Evol 8:9241–9258. https://doi.org/10.1002/ece3.4343
Oremus M, Poole MM, Steel D, Baker CS (2007) Isolation and interchange among insular spinner dolphin communities in the South Pacific revealed by individual identification and genetic diversity. Mar Ecol Prog Ser 336:275–289. https://doi.org/10.3354/meps336275
Palsbøll PJ, Zachariah Peery M, Bérubé M (2010) Detecting populations in the ‘ambiguous’ zone: kinship-based estimation of population structure at low genetic divergence. Mol Ecol Resour 10:797–805. https://doi.org/10.1111/j.1755-0998.2010.02887.x
Paquet M, Arlt D, Knape J et al (2020) Why we should care about movements: using spatially explicit integrated population models to assess habitat source–sink dynamics. J Anim Ecol 89:2922–2933. https://doi.org/10.1111/1365-2656.13357
Parsons KM, Noble LR, Reid RJ, Thompson PM (2002) Mitochondrial genetic diversity and population structuring of UK bottlenose dolphins (Tursiops truncatus): is the NE Scotland population demographically and geographically isolated? Biol Conserv 108:175–182. https://doi.org/10.1016/S0006-3207(02)00103-9
Parsons KM, Durban JW, Claridge DE et al (2006) Population genetic structure of coastal bottlenose dolphins (Tursiops truncatus) in the Northern Bahamas. Mar Mammal Sci 22:276–298. https://doi.org/10.1111/j.1748-7692.2006.00019.x
Peakall ROD, Smouse PE (2006) Genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Penteriani V, Ferrer M, Delgado MM (2011) Floater strategies and dynamics in birds, and their importance in conservation biology: towards an understanding of nonbreeders in avian populations. Anim Conserv 14:233–241. https://doi.org/10.1111/J.1469-1795.2010.00433.X
Pilot M, Dahlheim ME, Hoelzel AR (2010) Social cohesion among kin, gene flow without dispersal and the evolution of population genetic structure in the killer whale (Orcinus orca). J Evol Biol 23:20–31. https://doi.org/10.1111/j.1420-9101.2009.01887.x
Piroddi C, Bearzi G, Gonzalvo J, Christensen V (2011) From common to rare: the case of the Mediterranean common dolphin. Biol Conserv 144:2490–2498. https://doi.org/10.1016/j.biocon.2011.07.003
Pleslić G, Rako Gospić N, Mackelworth P et al (2015) The abundance of common bottlenose dolphins (Tursiops truncatus) in the former special marine reserve of the Cres-Lošinj Archipelago, Croatia. Aquat Conserv Mar Freshw Ecosyst 25:125–137. https://doi.org/10.1002/aqc.2416
Pleslić G, Rako-Gospić N, Miočić‐Stošić J et al (2019) Social structure and spatial distribution of bottlenose dolphins (Tursiops truncatus) along the Croatian Adriatic coast. Aquat Conserv Mar Freshw Ecosyst 29:2166–2132. https://doi.org/10.1002/aqc.3213
Pleslić G, Rako-Gospić N, Holcer D (2020) Bottlenose dolphins (Tursiops truncatus) in North Dalmatia, Croatia: occurrence and demographic parameters. Mar Mammal Sci 37:142–161. https://doi.org/10.1111/mms.12735
Pratt EAL, Beheregaray LB, Bilgmann K et al (2018) Hierarchical metapopulation structure in a highly mobile marine predator: the southern australian coastal bottlenose dolphin (Tursiops cf. australis). Conserv Genet 19:637–654. https://doi.org/10.1007/s10592-017-1043-6
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1093/genetics/155.2.945
Pulliam HR (1988) Sources, sinks, and Population Regulation. Am Nat 132:652–661
Rako N, Fortuna CM, Holcer D et al (2013) Leisure boating noise as a trigger for the displacement of the bottlenose dolphins of the Cres-Lošinj archipelago (northern Adriatic Sea, Croatia). Mar Pollut Bull 68:77–84. https://doi.org/10.1016/j.marpolbul.2012.12.019
Rako-Gospić N, Radulović M, Vučur T et al (2017) Factor associated variations in the home range of a resident Adriatic common bottlenose dolphin population. Mar Pollut Bull 124:234–244. https://doi.org/10.1016/j.marpolbul.2017.07.040
Rambaut A, Drummond AJ, Xie D et al (2018) Posterior summarization in bayesian phylogenetics using Tracer 1.7. Syst Biol 67:901–904. https://doi.org/10.1093/sysbio/syy032
Robinson KP, O’Brien JM, Berrow SD et al (2012) Discrete or not so discrete: long distance movements by coastal bottlenose dolphins in UK and Irish waters. J Cetacean Res Manag 12:365–371
Robles H, Ciudad C (2020) Assessing the buffer effect of floaters by reinforcing local colonization in spatially structured populations. Anim Conserv 23:484–490. https://doi.org/10.1111/acv.12566
Romanić SH, Holcer D, Lazar B et al (2014) Organochlorine contaminants in tissues of common bottlenose dolphins Tursiops truncatus from the northeastern part of the Adriatic Sea. Environ Toxicol Pharmacol 38:469–479. https://doi.org/10.1016/j.etap.2014.07.017
Rosel PE, Forgetta V, Dewar K (2005) Isolation and characterization of twelve polymorphic microsatellite markers in bottlenose dolphins (Tursiops truncatus). Mol Ecol Notes 5:830–833. https://doi.org/10.1111/j.1471-8286.2005.01078.x
Rosel PE, Hansen L, Hohn AA (2009) Restricted dispersal in a continuously distributed marine species: common bottlenose dolphins Tursiops truncatus in coastal waters of the western North Atlantic. Mol Ecol 18:5030–5045. https://doi.org/10.1111/j.1365-294X.2009.04413.x
Ryman N, Palm S (2006) POWSIM: a computer program for assessing statistical power when testing for genetic differentiation. Mol Ecol Notes 6:600–602. https://doi.org/10.1111/j.1471-8286.2006.01378.x
Sánchez-Montes G, Ariño AH, Vizmanos JL et al (2017) Effects of sample size and full sibs on genetic diversity characterization: a case study of three syntopic Iberian pond-breeding amphibians. J Hered 108:535–543. https://doi.org/10.1093/jhered/esx038
Sander N, Abel GJ, Bauer R, Schmidt J (2014) Visualising migration flow data with circular plots. The Vienna Institute of Demography Working Paper Series 02 / 2014, The Vienna Institute of Demography, Austrian Academy of Sciences, 35pp
Segura I, Rocha-Olivares A, Flores-Ramírez S, Rojas-Bracho L (2006) Conservation implications of the genetic and ecological distinction of Tursiops truncatus ecotypes in the Gulf of California. Biol Conserv 133:336–346. https://doi.org/10.1016/j.biocon.2006.06.017
Sellas AB, Wells RS, Rosel PE (2005) Mitochondrial and nuclear DNA analyses reveal fine scale geographic structure in bottlenose dolphins (Tursiops truncatus) in the Gulf of Mexico. Conserv Genet 6:715–728. https://doi.org/10.1007/s10592-005-9031-7
Smedbol RK, McPherson A, Hansen MM, Kenchington E (2002) Myths and moderation in marine metapopulations? Fish Fish 3:20–35. https://doi.org/10.1046/j.1467-2979.2002.00062.x
Viaud-Martinez K, Brownell R Jr, Komnenou A, Bohonak A (2008) Genetic isolation and morphological divergence of Black Sea bottlenose dolphins. Biol Conserv 141:1600–1611. https://doi.org/10.1016/j.biocon.2008.04.004
Wang J (2006) Informativeness of genetic markers for pairwise relationship and relatedness inference. Theor Popul Biol 70:300–321. https://doi.org/10.1016/J.TPB.2005.11.003
Wang J (2017) The computer program structure for assigning individuals to populations: easy to use but easier to misuse. Mol Ecol Resour 17:981–990. https://doi.org/10.1111/1755-0998.12650
Wang J (2019) A parsimony estimator of the number of populations from a STRUCTURE-like analysis. Mol Ecol Resour 19:970–981. https://doi.org/10.1111/1755-0998.13000
Waples RS, Gaggiotti OE (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439. https://doi.org/10.1111/j.1365-294X.2006.02890.x
Waples RS, Hoelzel AR, Gaggiotti O et al (2018) Guidelines for genetic data analysis. J Cetacean Res Manag 18:33–80
Wasser SK, Lundin JI, Ayres K et al (2017) Population growth is limited by nutritional impacts on pregnancy success in endangered Southern Resident killer whales (Orcinus orca). PLoS ONE 12:e0179824. https://doi.org/10.1371/journal.pone.0179824
Watson JR, Siegel DA, Kendall BE et al (2011) Identifying critical regions in small-world marine metapopulations. Proc Natl Acad Sci 108:E907–E913. https://doi.org/10.1073/PNAS.1111461108
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191. https://doi.org/10.1093/genetics/163.3.1177
Acknowledgements
This research was funded by the Italian DG Fishery within the research framework of the Italian obligations to the Council Regulation (EC) n. 812/2004 (BYCATCH programme) and by personal funding of Stefania Gaspari. The authors acknowledge the people that have provided samples: Morigenos - Slovenian Marine Mammal Society, Slovenia; Blue World Institute of Marine Research and Conservation, Croatia; Croatian Natural History Museum, Croatia; Tethys Research Institute, Italy; A.R.C.H.E. Porto Garibaldi, Italy (Carola Vallini); Università degli Studi di Siena, Italy (Letizia Marsili); University of Padoa, Italy (Bruno Cozzi, Maristella Giurisato). Work in the Gulf of Ambracia received logistic support from the Milan Civic Aquarium and Hydrobiological Station,Lefkas Marina and funding from OceanCare. We also thank Caterina Fortuna (ISPRA) and Antonello Sala (CNR IRBIM) for their support in carrying out this research.
Funding
Funding for laboratory genotyping work was provided by the Consiglio Nazionale delle Ricerche (CNR, Italy) BYCATCH research program.
Author information
Authors and Affiliations
Contributions
SG and AEM designed the study, supervised genetic data production, and wrote the first manuscript draft. CD, KS, CSS and NC carried out laboratory work. SG, AEM, CD and KS carried out data analyses. TG, JG and DH provided local expertise on resident bottlenose dolphin communities and local habitat conditions, contributed samples and critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Conflicts of interest/Competing interests
Authors declare no conflict of interests.
Ethics approval
Research approved by the University of Lincoln Ethics committee (CoSREC44).
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gaspari, S., Dooley, C., Shreves, K. et al. Connectivity patterns of bottlenose dolphins (Tursiops truncatus) in the north-east Mediterranean: implications for local conservation. Conserv Genet 25, 409–426 (2024). https://doi.org/10.1007/s10592-023-01577-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-023-01577-4