
Abstract
Genetic diversity of organisms is an indicator of their long-term survival and can potentially be shaped by the extent of geneflow between populations. Geographical features and anthropogenic interferences can both obstruct and also facilitate animal movement, directly or indirectly. Such patterns have not been extensively studied across grasslands in the Indian subcontinent which is a mosaic of both natural and man-made topography. This study looks at genetic variation in an endemic ungulate, the Antilope cervicapra or blackbuck, throughout its distribution range. Using mitochondrial and nuclear (microsatellite) information, we find that different markers shed light on different aspects of their evolutionary history. Absence of robust geographical clustering in mitochondrial DNA indicate recent isolation in these populations, while lack of shared haplotypes between sampling locations suggests female philopatry. Nuclear data shows the presence of three genetic clusters in this species, pertaining to the Northern, Southern and Eastern regions of India. Our study also shows that an ancestral stock separated into two groups that gave rise to the North and South clusters and the East population was derived from the South at a later time period. Both microsatellite and mitochondrial data indicate that the population from the Eastern part of India is genetically distinct and the species as a whole shows signatures of having undergone recent genetic expansion. In spite of immense losses in grassland habitats across India, blackbucks seem to have well-adapted to human altered landscapes and their numbers are beginning to show an upward trend.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
A widely recognized approach towards characterizing diversity is studying and comparing genetic variation within and between life forms (Noss 1990). Considering the necessity of genetic diversity for evolution to occur (Reed and Frankham 2003), it is often used as a measure of fitness and to determine long-term viability of organisms. The genetic variability of an organism allows us to assess its ability to respond to changing environmental conditions or disease epidemics (Rasmussen et al. 2014; Rivers et al. 2014; Hume et al. 2016). The maintenance of genetic diversity is largely shaped by population size and geneflow between populations (Frankel and Soule 1981; Frankham 1996) with larger connected populations showing higher genetic diversity (Futuyma 1986; Falconer 1996). In this regard, extrinsic factors such as habitat continuity and natural barriers play an important role in shaping genetic diversity. Depending on the species’ traits, relief features like mountains, forests, dry grasslands, river basins etc., may hinder movement (Roonwal 1984; Knowles 2000; Nag et al. 2011) or facilitate connections (Willis et al. 2010) or even act as refugia (Ripley and Beehler 1990; Modolo et al. 2005) during unfavourable conditions, hence modifying patterns of geneflow and leading to genetic structure. In recent times, anthropogenic factors such as loss and/ or fragmentation of habitats have also reduced connectivity and fostered genetic structure, as well as loss of genetic diversity. The combination of limited geneflow and small size could cause genetic differences to accumulate rapidly between geographically isolated populations (Worley et al. 2004). In the context of habitat fragmentation, knowledge about how diversity is partitioned within and among populations is important for species-centric conservation programs. Small patches separated by stringent barriers, natural or human-made, could lead to distinct populations or even sub-species on either side of the barrier. Additionally, it could also lead to loss of effective population size, or bottleneck effects (Lukoschek 2018; Frankham et al. 2002) which could be inferred from their underlying genetic condition. Further, using markers with a higher mutation rate can provide information on recent population genetic processes (Wang et al. 2019). This knowledge could be used to identify sources of introduced populations (Goodman et al. 2001), the most vulnerable populations requiring immediate action, and also, genetically diverse populations which can recover them.
The Indian sub-continent is a prime example of a mosaic landscape, with a complex ecological, climatic and geological history (Biswas and Pawar 2006) and harbouring diverse ecosystems criss-crossed with topological barriers as well patches of human-altered regions, that shelter an astounding biodiversity (Ghosh-Harihar et al. 2019). However, not many studies in India have looked at the impact of landscape alteration and habitat fragmentation on the genetic diversity of a species, especially across its entire range. Additionally, we do not have a complete understanding of regional biodiversity patterns in India (Reddy 2014; Tamma and Ramakrishnan 2015). This is even more so in cases of meso or mega herbivore fauna, particularly in grassland ecosystems. Few findings indicate the possibility of both hitherto unknown genetic connectivity as well as barriers to geneflow, depending on the species under study. Research on tiger populations from six protected areas in central India showed genetic connectivity between these populations, in spite of the presence of seemingly in hospitable regions in between (Joshi et al. 2013). On the other hand, leopard cats from the North and South of India have a high population structure, indicating a climatic barrier (Mukherjee et al. 2010). A leopard meta-population in central India shows genetic structuring due to increasing habitat fragmentation, both at the landscape and fine-scale levels (Dutta et al. 2013). Elephant populations in Southern India belong to two genetic clusters, corresponding to the Nilgiri and Periyar-Anamalai regions, and these are further distinct from those in central and northern India (Vidya et al. 2005a, b). While sloth bears in the Satpura-Maikal region of central India separate into two genetic clusters, there is also evidence of contemporary geneflow (Dutta et al. 2015). All of these examples come from investigations on forest dwelling species while the dry-habitat ones have been largely ignored. The same topographic and climatic barriers would affect the distribution of arid-zone species in a different manner, in addition to the challenges presented by habitat fragmentation. The Indian grey wolves also inhabit semi-arid grasslands and scrub forests (Jhala and Giles 1991) and their phylogeography has been well studied (Sharma et al. 2004), albeit with a distinct lack of samples from southern India. Another species that could potentially display strong genetic structure is the grassland dwelling blackbuck or Antilope cervicapra, an endemic antelope from the Indian sub-continent. They are phylogenetically nested within the gazelle clade (Jana and Karanth 2019), yet are morphologically distinct and sexually dimorphic. They are medium sized antelopes (23–45 kg; Mungall 1978; Ranjitsinh 1989) and form a part of the Bovidae family. They are selective grazers that live in groups which can range from two to several hundred individuals (Ranjitsingh 1989). While places like Velavadar in Gujarat and Tal Chhappar in Rajasthan boast of the largest natural blackbuck populations, we also find small groups of less than ten in other places like Kolar in Karnataka or Ananthapur district in Andhra Pradesh (personal observations). They are seen in isolated clusters and also in seemingly connected metapopulations, across different parts of the country, making it an ideal system to study the different patterns of genetic structure and variability and also, demographic history in the context of habitat fragmentation. Blackbucks have never been found, even as fossils, outside the sub-continent, although they had been introduced in North America and Argentina, for game hunting. Once prevalent across India, their numbers were greatly decimated in the past due to indiscriminate poaching and game hunting. Post that period, shrinkage and fragmentation of grasslands to relic pockets further stained these isolated populations (Schaller 2009). Currently, very few patches of grasslands and scrublands remain relatively undisturbed across the country and these areas continue to be relegated as wastelands and are often considered as prime targets for development, urbanisation and even land conversion for agriculture. They are currently seen in multiple areas across the semi-arid regions in India. Given that grasslands in India continue to be heavily fragmented, it is imperative to understand the population structure of blackbucks and make effective decisions regarding their conservation.
Most of our information on antelopes comes from African species. Asia has fewer antelopes, but they are found in a wider range of habitats (Mallon and Kingswood 2001). Non-invasive methods offer us a way for conducting genetic analysis of species that may be difficult to sample otherwise. Methods like using fecal samples allow us to obtain genetic information for animals without having to euthanize, maim or even capture the focal samples, earning approval from both conservation studies and ethical perspectives. However, the extracted DNA is often highly fragmented, of poor quality and quantity and includes microbial and dietary genetic material, making downstream experiments and analysis more difficult. A fraction of collected samples that do not pass the quality threshold have to be discarded, resulting in reduced sample sizes. Some strategies that can support more reliable results include repeated genotyping of samples to ensure accuracy (Taberlet et al. 1996) and rejection of samples containing a lower amount of DNA than a reliability threshold (Morin 2001; Horvath et al. 2005).
Here we study how genetic variation is distributed across the range of a native Asian grassland specific antelope, the blackbuck. While previous work has reported the forensic identification of blackbucks for wildlife investigations (Kumar et al. 2018), two very recent studies have begun advancing our understanding of the genetic diversity in this species. Bhaskar et al., utilised three mitochondrial markers covering cytochrome b, cytochrome c oxidase subunit-1 and the partial control region to study multiple populations in southern India and found evidence of higher than usual haplotype diversity and some genetic structure between three clusters. De et al., sampled a single region from northern India and were able to cross-amplify a panel of ungulate microsatellite markers which could be potentially used for population and landscape genetics studies of the blackbuck. However, we still lack a comprehensive knowledge of their genetic diversity, involving different markers, multilocus analysis and well as extensive sampling covering their pan-India geographical distribution. To elucidate the patterns of genetic structure within this species we used both nuclear (microsatellite) and mitochondrial (D-loop) markers. Furthermore, we also explore the drivers of genetic variation and make inferences on past demography in this species.
Materials and methods
Sampling
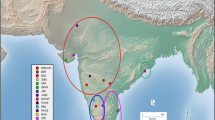
Fecal samples of blackbucks were collected non-invasively, without animal-handling from 12 different locations (map in Fig. 1) across their geographic range, in the states of Rajasthan, Gujarat, Maharashtra, Karnataka, Tamil Nadu, Andhra Pradesh, Uttar Pradesh and Orissa (details in Table 1). In case of protected areas, permissions to conduct research were obtained from the requisite forest departments. The animals were observed from a distance and tracked on foot or followed on vehicles. Being diurnal, blackbucks spend most of their time foraging during early morning and afternoon and hence, most of the sampling was conducted in this period. Population densities of blackbucks at each sampling location were obtained from either literature, previous surveys or forest department records, where available (Bikash Rath and Rao 2005; Asif and Modse 2016; Baskaran et al. 2016; Mamatha and Hosetti 2018; Meena and Saran 2018; BirdLife International, 2020). This was used as a proxy for population sizes for the purpose of our study, since we had access to more accurate records of densities rather than actual number of individuals in a particular location. Since blackbuck fecal pellets have a characteristic fecal shape (Bhaskar et al. 2021), they are easily distinguishable from those of other bovid species from that region, thus negating any potential mix-up. Fresh fecal samples from individual adult blackbucks were collected using two methods. First, the outer layer of the fecal pellets was swabbed and preserved in a lysis buffer solution (White and Densmore 1992) in 2.0 ml cryovials, to prevent the DNA from degradation. The buffer was prepared using 2.5 g of Sodium dodecyl sulphate (SDS), 100 ml of 0.5 M Ethylene diamine tetra-acetic acid (EDTA), 1ml Sodium chloride (NaCl) and 50 ml of 1 M Tris-Hydrochloric acid (HCl) and milliQ water to make up the volume to 500 ml. Secondly, the whole pellets were collected and stored in absolute alcohol. The samples were stored at room temperature in the field and DNA extraction was carried out after they were transferred to the laboratory.

Map showing the different sampling locations. The populations were grouped into East (Green), North (Red) and South (Blue) clusters and are colour coded as per the labels of the haplotype network. Inset: Male adult blackbuck
Molecular work
The DNA was extracted from the swabbed samples using the Wizard Genomic DNA Purification kit (Promega, Singapore) and from the whole pellets using the QIAmp Fast Stool Extraction kits (Qiagen, Germany), following the standard protocols, with certain modifications. Following the first step, where the outer layer of the pellets was scraped into fine powder, the samples were kept in extraction buffer overnight, before proceeding to the next steps. The DNA extracts were quality checked using a nanodrop, aliquoted and diluted about five-fold, and stored at − 20 °C. For the mitochondrial marker, the D-loop region sequence was obtained from the complete mitochondrial genome of A. cervicapra (GenBank Accession number: AP003422.1). The region was divided into four overlapping segments which acted as templates to design primers using the web tool Primer-BLAST. The default primer stringency conditions were used and the primer melting temperature (Tm) range was set from 45 °C to 60 °C, with a maximum Tm difference of 3 degrees. Primer pairs were selected from the resultant options on the basis of maximum coverage and higher GC content. Following preliminary trials, four overlapping primers viz., HV1D, INT, DLF3 and DLF4 (Table 2), covering < 300 base pairs each, were finalized to amplify the D-loop region spanning ~ 800 base pairs. Double stranded polymerase chain reactions (PCRs) were performed using ~ 5–20 ng/ul of the extracted DNA, with 1U Taq Polymerase, 2 mM of Magnesium chloride (MgCl2), 0.15 mM of deoxynucleotide triphosphates (dNTPs), 2ng/ml of Bovine Serum Albumin (BSA), 10 μm forward and reverse primers and milliQ water to make up a final reaction volume of 10ul, using the following thermocycler settings: 94 °C initial temperature (5 min), 45–50 cycles of 94 °C denaturation (30 s), 45–63 °C annealing (30 s), 72 °C extension (1 min) and 72 °C final extension (10 min). The PCR products were run on 1% agarose gel using electrophoresis and observed under ultraviolet light to select for successful amplification and the Sanger sequencing was outsourced to Medauxin and Barcode Biosciences. Each sample was sequenced in both forward and reverse directions and the obtained sequence was aligned using the Genbank data to avoid potential Numts and ensure the validity of the blackbuck mitochondrial DNA region. The sequences for unique haplotypes were submitted to Genbank (Accession numbers: OP794109-OP794335).
Due to the unavailability of published primers for microsatellite loci in blackbucks, primers from related species were tested in randomly selected samples. Of the 25 bovid primer pairs selected, bm302, bm415, maf70, hdz496, inra040, tgla122 and sps115 (Table 2), showed successful amplification during the preliminary trials and were further used for genotyping. The PCRs were performed using ~ 5–20 ng/ul of the extracted DNA as template, with 1x Qiagen Multiplex master mix, 10 μm of fluorescence-labelled forward (FAM or 6HEX) and reverse primers, 2 ng/ml of BSA and milliQ water to make up a final volume of 10ul, using the following thermocycler settings: 94 °C initial temperature (5 min), 50–55 cycles of 94 °C denaturation (30 s), 50–60 °C annealing (30 s), 72 °C extension (1 min) and 72 °C final extension (10 min). In light of suggested strategies to minimize genotyping errors (mentioned earlier in Introduction), each sample was run at least 3 times to ascertain consistent results for minimising scoring errors and also to verify the final alleles called. The PCR products were outsourced to Barcode Biosciences for genotyping, using LIZ500 as a size standard. Care was taken to avoid exposure to direct light during setting up the reactions, to prevent degradation of the fluorescent-labelled primers.
Analyses
Mitochondrial markers
The sequences obtained were edited using Chromas v2.6.5 (technelysium.com.au/chromas.html), to correct erroneous base calling. The cleaned sequences were then aligned using the Muscle algorithm with default parameters in Mega v7 (Tamura et al. 2011) and the four overlapping sequences obtained for each individual were combined using Mega and Geneious v10.1 (Kearse et al. 2012). The nucleotide and haplotype diversity values for each of the sampling locations were calculated using DnaSP v6.11.01 (Rozas et al. 2017). The concatenated dataset (> 800 bps), containing only the complete sequences obtained (without missing information), was used to calculate the haplotype diversity and to generate a Roehl data file in DnaSP. This was then used to build a build a median joining haplotype network in NETWORK v5.1 (fluxus-engineering.com), without star contraction and using the default values for epsilon. The original median joining network was post-processed on the basis of maximum parsimony using the Steiner algorithm (Polzin and Daneshmand 2001), to reduce the number of loops crossing over and obtain a network that could be visualised better. Further, the entire dataset assuming a single population was also used to calculate Tajima’s D and Fu’s FS values in Arlequin v3.5.2.2 (Excoffier and Lischer 2010). The mitochondrial sequences were segregated into three clusters viz., North (N), East (E) and South (S) based on their geographical location (Table 1) and used for an analysis of molecular variation (AMOVA) in Arlequin v3.5.2.2, to compare the genetic differences between clusters and also quantify the percentage of genetic variation explained within and between clusters. Further, a paired Mantel test was performed for the pairwise genetic and geographic distances between samples to check for isolation by distance (IBD).
Microsatellite data
For microsatellite screening, the selected 25 loci were used for amplification in > 25 samples, and included members from all the sampling locations. Most of the loci did not show any PCR amplification, even under multiple cycling conditions. The final loci to use for analysis were selected by using samples that showed a clear amplification in > 50% of the samples belonging to different sampling locations.
Each locus was amplified and scored at least three times (in each sample), in order to minimize genotyping errors. The samples that did not show successful amplification or consistent peaks in three genotyping replicates were discarded and the estimation of null alleles and allelic dropout, along with checking for scoring errors was done using MICROCHECKER v2.2.3 (Van Oosterhout et al. 2004). The genotyping results were viewed using the microsatellite plug-in in Geneious v10.1, to identify the size standard and the allele calling was done manually, post visualising the microsatellite stutter peaks. The Pid/PI (probability of misidentifying two individuals, drawn from the same randomly mating population, as a single individual) and Pidsibs/PIsibs (misidentifying siblings as the same individual, taking into account genetic similarity) values for each of the population was calculated separately, both at each locus and cumulatively using GenAlEx v6.503 (Peakall and Smouse 2006, 2012) in Microsoft Excel 2016. Further, the genetic diversity was measured as the number of alleles per population (Na), population-wise unbiased expected (He) (Nei 1978) and observed (Ho) heterozygosity, and tested for Hardy Weinberg equilibrium (HWE). The complete microsatellite dataset (without considering location information) was also used to calculate overall observed and expected heterozygosities and deviations from Hardy Weinberg equilibrium and linkage disequlibrium in Arlequin v3.5.2.2. The allele size information from each of the loci was also used to build a pairwise distance matrix (unbiased Nei’s distance) between individuals, which was then used for a principal coordinates analysis (PCoA) and to build a neighbour joining tree in MEGA. Further, the pairwise FST values between populations was used for a paired Mantel test with pairwise geographic distances in GenAlEx to check for isolation by distance.
Genetic clusters were determined using a Bayesian clustering approach in STRUCTURE v2.3.4 software (Pritchard et al. 2000), to determine whether there was any hidden population structure, irrespective of the geographical location of samples. The software can be used to group individuals into K clusters based on their genotypes, without prior information on their geographical location, using a Markov Chain Monte Carlo (MCMC) approach. An admixture model was used, with a Dirichlet prior D, where the relative contributions of the K populations were modelled using K parameters. A uniform prior with a maximum of 10.0 was used and the initial alpha value was set at 1.0 and standard deviation (SD) to 0.025. Allele frequencies were considered to be correlated among populations and FST value were considered ‘different for different subpopulations’, with prior mean of 0.01 and SD of 0.05. The analysis was run for K values between one and twelve and 20 runs were performed for each K with 10,000 iterations following a burn-in of 1000 steps. The results were used as input for structure harvester to determine the optimal number of genetic clusters using the method by Evanno et al. (2005), based on estimation of the mean likelihood value per K.
A simulation-based approach was used on the whole population to test for departure from mutation-drift equilibrium and detect signatures of past population dynamics. Two mutation models, the single stepwise model (SSM) and the two-phase model (TPM) were used and the Wilcoxon sign-rank test was implemented in BOTTLENECK v1.2.02 (Cornuet and Luikart 1996). The TPM model was run using both, the default variance of the geometric distribution and proportion of SMM in TPM values and also with variance = 0.36 and proportion = 0.0 as suggested by the authors, as they correspond to sensible parameter values for most microsatellites. The analysis was run for the species, from all sampling locations across its range, using the entire dataset as a single population.
The nucleotide diversity obtained from the mitochondrial data and the Ho and Na values obtained using the microsatellite markers were used as proxies for genetic diversity and compared against the population densities at each of the sampling locations. A linear regression was performed to determine whether there was an association between population density and genetic diversity and an analysis of variance (ANOVA) to test whether the correlations were significant.
Historical demography
Finally, to understand past demographic changes in the populations of this species, an Approximate Bayesian Computation was implemented on the microsatellite dataset using DIYABC v2.0 (Cornuet et al. 2014). The three geographic clusters, North, South and East were coded as populations and three demographic scenarios (Fig. 5) were compared after computing 3 million simulated datasets. In the first scenario, the South cluster was derived from the North (at time t2), which harboured the ancestral population, and the East diverged from the South at a later time period (t1). It is known that dispersal of the Bovidae family into the Indian subcontinent occurred predominantly through the Northwestern faunal gateway (Kurup 1974). The ancestral range of antelopes being the Saharo-Arabian region (comprising of Northern Africa and the middle East), the antelope lineages in India are presumed to have followed a similar route (Jana and Karanth 2019). In the second scenario, the South cluster was more ancient, and the North and East clusters were derived from it at successive time periods (t2 and t1 respectively). This also accounts for the possibility that the Southern region served as a ‘refugia’ during the Pleistocene (Vidya et al. 2009), from which the other two clusters diverged more recently. In the third scenario, the ancestral population (NA) gave rise to the North and South clusters (at time t2) and the East cluster was derived from the South at a later period (t1). The scenario and parameter prior combinations were first pre-evaluated using a Principal Component Analysis (PCA). This was done to check if a significant proportion of the simulated dataset was different from the values in the observed dataset. The stepwise mutation model was used, and the posterior probabilities of the scenarios was computed to find the most likely demographic pattern. The Bayesian model choice was made more efficient by using linear discriminant analysis on summary statistics, as suggested by the authors. Each of the scenarios was individually tested using both direct estimate and logistic regression to compare the number of selected datasets closets to the observed dataset. The confidence in scenario choice was evaluated using posterior based errors (where test samples were drawn from the simulated datasets closest to the observed dataset) and prior based errors (test samples randomly drawn from scenario ID and parameter values in prior distributions) when computed globally over all scenarios. Further, the chosen scenario was separately checked for prior error rate when compared against all the other scenarios in consideration, to deduce any type I errors.

Haplotype network of all samples showing unique haplotypes for all locations
Results
Mitochondrial dataset
About 400 individuals were sampled for both, the mitochondrial and microsatellite analysis and ~ 50% of them showed successful amplification in each case. Since extraction from fecal samples yielded low quality, fragmented DNA, separate PCRs were performed to amplify four smaller, over-lapping regions of the D-loop, that were then concatenated. Unambiguous results from each sequencing reaction were selected and the final dataset consisted of 810 bps D-loop sequence from 227 individuals. The three samples from Jayamangali did not show amplification for the first ~ 300 bps region of the D-loop, which also contains the hypervariable region 1. Hence, these samples were removed from the dataset used for building the haplotype network in NETWORK. A total of 186 haplotypes were found in the dataset and the network showed distinct haplotypes from all sampling locations, however, it did not exhibit any geographical clustering of haplotypes (Fig. 2). An overall pattern of isolation by distance was detected across the study region by the Mantel test (r = 0.25, p < 0.01).

Neighbour joining tree built using pairwise genetic distance between all samples, from 7 microsatellite loci. The highlighted region shows the clusters where samples from Bhetanai (East) are present. The sample labels are colour coded according to the North (red), South (blue) and East (green) clusters
The analysis of molecular variance (AMOVA) showed that 89.52% of the variation was explained within the three geographic clusters, North, South and East and only 10. 48% was sourced/contributed from between clusters. These geographic clusters were delimited based on the STRUCTURE analyses of microsatellite data (see below). The Mantel For the entire species, the haplotype diversity was high (0.997) while the nucleotide diversity was low (0.0667). Tajima’s D was − 1.474 and Fu’s FS was − 23.877 and both values were significantly negative. A comparison using pairwise FST values between clusters showed a 9.57% difference between the North and South cluster, 10% difference between the South and East and 13.45% difference between North and East clusters and the FST p values were all significant (p < < 0.01). The nucleotide diversity from mitochondrial D-loop data showed a significant correlation with the population densities at the sampling locations (F = 19.05, p < 0.01), with the linear model having an adjusted R2 of 0.6673. We found that mitochondrial genetic diversity was positively associated with blackbuck density (t = 4.365, p = 0.0024).
Microsatellite dataset
In our experiments, only seven of the tested loci showed successful amplification. Successful amplification of microsatellites markers from fecal samples is effected by many factors, including climatic conditions, sample preservation, time between collection and DNA extraction, PCR inhibitors, improper annealing, low DNA template, among others. Other studies have also shown that a large proportion of markers screened for blackbuck could not be amplified, indicating mutated or non-conserved flanking regions of the primer binding sequences (De et al. 2021). Large allelic dropout and the presence of null alleles also hinder obtaining accurate data, making it challenging to obtain a large panel of microsatellite markers for diversity-based statistical analysis. Further, genotyping errors can frequently occur while working with microsatellite markers, especially when working with non-invasive genetic samples. These may lead to a positive bias, which accumulates proportionally with increasing number of markers used (Creel et al. 2003). We were able to obtain a final dataset of 213 individuals from the sampled 400, which showed consistent peaks in the genotyping results. MICROCHECKER did not show evidence of large allelic dropout in any of the seven loci. Null alleles were indicated in the sampling sites (ranging from one to six loci) although they were inferred as a result of homozygote excess in each case. The repeat regions could not be successfully amplified for most of the samples from Challakere and hence, this location was dropped from further microsatellite analyses (Table 1). All the seven loci were polymorphic, with 8–20 alleles each and the mean number of alleles ranged from 3.143 to 7.714 per site and did not depend on the population density at those sites (linear regression: r = 0.505, p > > 0.05). All loci showed an overall deviation from HW equilibrium although, among all the 77 possible locus-site combinations (7 loci × 11 populations), 34 did not show this pattern (p > 0.01). The expected heterozygosity values ranged from 0.509 to 0.730 and the observed heterozygosities were lower (0.256–0.579). The microsatellite heterozygosities did not show any significant correlation to the population densities from sampling locations (p > > 0.05), unlike nucleotide diversity from mtDNA. The cumulative Pid/PI was < 0.000001 in most cases with the exception of Kolar (0.0001), corresponding to < < 0.001% probability that two individuals may have been mistakenly considered as one, and a < 0.01% chance of two siblings misidentified and a single blackbuck. The cumulative Pid/PI and Pidsibs/PIsibs values indicated a min of 5 microsatellites loci were required to allow for robust discrimination between two blackbucks (PID < 0.001 and PID sib < 0.01).
The Neighbour joining tree built (Fig. 3) did not show any particular structure for most sampling locations, except for Bhetanai, the majority of which fell in a single cluster (with the exception of four samples, which were closer to other populations). This was further supported by the PCoA analysis for blackbucks across eleven sites. The first axis explained 10.95% of variation while the first three axes combined explained 26.7% of the variation (Supplementary fig. S1). The Mantel test did not show any correlation between the pairwise FST values between sampling locations and their geographical distances (r < < 0.001, p > > 0.05), indicating no isolation by distance.

a Graph showing the DeltaK values for each K assigned in STRUCTURE. b Bar plot of STRUCTURE analyses arranged according the three clusters (K = 3)
The results from STRUCTURE and Structure Harvester showed K = 3 as the optimum value with the highest likelihood (Delta K = 29.946), indicating 3 most likely partitions (Fig. 4). Cluster 1 (C1, Eastern Cluster) almost completely contained individuals from Bhetanai, whereas cluster 2 (C2, Northern Cluster) is mostly composed of individuals from Velavadar, Tal Chhappar, Uttar Pradesh and Jayamangali and cluster 3 (C3, Southern Cluster) majorly from Rollapadu, Point Calimere, Kolar and Ranebennur. Samples from Nannaj were almost evenly distributed between clusters 2 and 3 (9 in C2 and 6 in C3) and two thirds of the animals from Timbaktu belonged to C3 and the rest in C2 (details of sampling locations in Supplementary Fig. S2).

Historical demographic scenarios tested using DIYABC. The green, red and blue regions correspond to the East (Pop1), North (Pop2) and South (Pop3) respectively. Results indicated Scenario 3 as the most likely scenario (See supplementary figure S3)
The Wilcoxon rank sign test using the BOTTLENECK software showed significant heterozygote deficiency in both, when SMM (p = 0.012) and TPM (p = 0.027) models (with suggested parameters for microsatellites) were assumed.
Historical demography
The first two axes of the PCA plot generated using DIYABC explained more than 55% of the variation. The observed dataset was within the simulated dataset space and most of the summary statistics values for the two datasets were not significantly different. Both the direct estimate and logistic regression considered scenario 3 as the best historical model of demographic change, by a considerable margin (Fig. 5; Supplementary Fig. S3). In the third scenario, the ancestral population gave rise to the North and South clusters and the East cluster was derived from the South at a later period. Further evaluation of confidence in our scenario choice also showed a high probability of type I error, where scenario 3 was rejected even though it was the correct choice, giving more validity to the results.
Discussion
Habitat loss and fragmentation has been known to influence genetic diversity in many organisms, although the extent of such affects has not been studied in grassland ecosystems in India. Here, we focus on an endemic antelope, to elucidate the patterns of genetic structure within this species, and also make inferences on past demography, using both nuclear (microsatellite) and mitochondrial markers. Since we were interested in both phylogeography and population genetics of the species, we put greater focus on maximised coverage of the geographical distribution range of the blackbucks. Although we were able to collect ~ 400 samples from our fieldwork, there were logistical constraints in larger sample collection, immediate DNA extraction and complete mitochondrial D-loop amplification. Non-invasive genetic studies are known to suffer from poor DNA quality and quantity (ref) and one way of ensuring accuracy of the results is by discarding samples below a certain reliability threshold, Our final tally of ~ 230 reported in this study belonged to samples that showed robust amplification across the complete mitochondrial D-loop and well as successful and consistent genotyping in three replicates. Though the study uses a small set of microsatellite markers, the combination of 7 loci showed sufficient resolution for correctly distinguishing between separate individuals. Since, population genetic parameters and frequency-based diversity analysis are less effected by genotyping errors as compared to individual identification (Pompanon et al. 2005), the seven selected loci in our study should have sufficient resolution for further genetic analysis of the focal species.
The mitochondrial haplotype network did not show any geographical clustering although each sampling location had unique haplotypes. The lack of shared haplotypes even between blackbucks from closely located regions suggests restricted female dispersal (Kerth et al. 2000; Lappan 2007). This is also supported by the fact that we find signatures of isolation by distance in maternally inherited mitochondrial DNA but not in case of microsatellite data, suggesting that male blackbucks might be the dispersing sex in this species. Many mammals are known to exhibit similar behaviour on account of female philopatry or male-biased dispersal and a polygynous mating system (Greenwood 1980; Clutton-Brock 1989; Waterman 2008; Nutt 2008).
The Neighbour joining tree generated using microsatellite data also did not exhibit geographical clustering of samples, with one notable exception. The majority of samples from Bhetanai (which is also the East cluster) fall in a single cluster, and this was also highlighted by the PCoA which shows notable separation between samples from Bhetanai and the other sites. The Bayesian method implemented in Structure supported three optimal clusters, one of which primarily contained samples from the East (Bhetanai) and the other two were mostly consisted of samples from the North (Velavadar, Tal Chhappar and Haliya) and South (Rollapadu, Point Calimere, Ranebennur, Kolar, Timbaktu). When these geographic clusters were delineated in the mitochondrial data for comparison, we found that a small, albeit significant, percentage of the genetic variation was explained between clusters.
Although the microsatellite diversity indices did not show any significant statistical correlation with the population densities, the mitochondrial nucleotide diversities of the sampled regions were positively correlated, with larger populations also showing higher mitochondrial genetic diversity. This again could potentially be a result of greater dispersal of male blackbucks and hence greater geneflow, when considering nuclear markers. In contrast, restricted movement of blackbuck females coupled with smaller effective population size of mtDNA marker (Moore 1995) may have contributed to the observed correlation between mitochondrial nucleotide diversities and population density.
While the mitochondrial haplotype network does not support geographic clustering of haplotypes, on the other hand, we find some genetic clustering into three populations in the microsatellite data. The simulation based approximated Bayesian computation strongly supported a past demographic scenario where an ancestral population gave rise to two genetic clusters, that pertain to populations in the North and South. The third population diverged from the South cluster at a later time period that gave rise to the current population in the East. These clusters are congruent with potential geographical barriers to species distribution and movement (Mani 1974; Ripley and Beehler 1990). Peninsular India is separated from northern India by a host of hill ranges prominently among them, the Satpura range, flanked by the Vindhyan and Ajanta hills (Thangaraj et al. 2010). Further, the Narmada and Tapti river basins also feature among prominent barriers (Thangaraj et al. 2010; Ramachandran et al. 2017). Northern India also experiences longer periods of extreme temperature, both in summer and winter when compared to much of peninsular India (Roy 2019). The two bifurcated regions correspond to the locations encompassing the South and North clusters mentioned in our study. The East cluster, although falling within peninsular India, is also isolated by forests and hill ranges belonging to the northern part of Eastern Ghats, as well as the Mahanadi and the Godavari and Krishna river basins on either side (Ramachandran et al. 2017). The highly fragmented series of hills in the southern Eastern Ghats may not pose as strong a barrier to the Point Calimere population, which is also located on the East Coast and forms one of the southernmost populations of this species. While most of these relief features have been considered as refugia or stepping-stones for wet-zone elements that aided their expansion (Hora 1949), the same topology could have also disrupted the movement of dry-zone species, even if they did not form a stringent barrier. However, it must be noted that signatures of historical demography could be distorted by recent geneflow. In case of blackbucks, our molecular data does not support strong population genetic structure, suggesting the possibility of such recent geneflow. Hence, the demographic scenario proposed here needs further validation.
From BOTTLENECK analysis on the entire species across its range, the SMM and TPM models showed significant heterozygote deficiency, which is indicative of a recent population growth (Cornuet and Luikart 1996; Sonsthagen et al. 2017). From the mitochondrial data, the significance of both, negative Tajima’s D (Tajima 1996) and Fu’s FS (Fu 1997), as well presence of a low nucleotide but high haplotype diversity, also points to recent expansion in this species. Large numbers of blackbucks were killed in the pre-independence (before 1947) era in India (Hughes 2009; Gilmour 2018) and poaching continued to be prevalent thereafter, resulting in precipitous decline in populations warranting threatened status for this species (Mallon 2008). However, recent surveys indicate a rise in numbers (IUCN SSC Antelope Specialist Group 2017). The protection accorded to this species under the Wildlife Protection Act in 1972 might have helped stabilize blackbuck populations. Furthermore, blackbucks seem to be well adapted to woodlands, scrublands, agricultural fields and even plantations. Thus, a combination of better protection and adaptability of this species to human modified landscapes probably aided the demographic expansion of the species in recent times, in spite of continued loss and fragmentation of grasslands brought about by anthropogenic activities. The demographic expansion in turn might have resulted in the lack of mitochondrial and nuclear population genetic structure (Excoffier et al. 2009; Garcia-Cisneros et al. 2016; Sabuni et al. 2016; Wereszczuk et al. 2017). However, much of the geneflow appears to be male mediated and it remains to be seen how restricted female movement might shape the demography of this species in the future.
Conclusion
Our study throws light on the genetic diversity of an endemic antelope species across its range and uncovers certain interesting patterns. Males of this species seem to move about a lot more as compared to females, as indicated by isolation by distance in mitochondrial but not nuclear markers, along with unique mitochondrial haplotypes from each region. We find the existence of three genetic clusters that coincide with current geographic regions, potentially facilitated by a host of biogeographic barriers in the Indian subcontinent. Our results indicate that a single ancestral population gave rise to blackbuck populations in the North and South, while the East population arose more recently from the South and is more distinct than the rest. Recent population expansion in A cervicapra as observed from both nuclear and mitochondrial analyses, as well as their increasing adaptation to human-modified landscapes point to the stability of this species, renewing hopes for their long-term survival. This study adds to the increasing body of work on phylogeography in the Indian subcontinent and tells us how both historic (geographic) and current anthropogenic processes might be shaping the demography of species in tandem. Similar future research that considers genetic markers with different evolutionary histories would give us a clearer overall picture and help in greenlighting strategies for conservation, especially in the case of endangered or vulnerable species.
Code availability
Not applicable.
Data availability
The DNA sequences used in this study have been uploaded to GenBank.
References
Al-Atiyat RM, Salameh NM, & Tabbaa MJ (2014) Analysis of genetic diversity and differentiation of sheep populations in Jordan. Electron J Biotechnol 17(4): 168–173
Asif M, Modse S (2016) The distribution pattern and population of blackbuck Antilope cervicapra Linnaeus in Bidar, Karnataka. Indian For 142:965–970
Baskaran N, Ramkumaran K, Karthikeyan G (2016) Spatial and dietary overlap between blackbuck (Antilope cervicapra) and feral horse (Equus caballus) at Point Calimere Wildlife Sanctuary, Southern India: competition between native versus introduced species. Mamm Biol. https://doi.org/10.1016/j.mambio.2016.02.004
Beja‐Pereira ALBANO, Zeyl EVE, Ouragh L, Nagash H, Ferrand N, Taberlet P, Luikart G (2004) Twenty polymorphic microsatellites in two of North Africa’s most threatened ungulates: Gazella dorcas and Ammotragus lervia (Bovidae; Artiodactyla). Mol Ecol Notes 4(3):452–455
Bhaskar R, Kanaparthi P, Sakthivel R (2021) Genetic diversity and phylogenetic analysis of blackbuck (Antilope cervicapra) in southern India. Mol Biol Rep 48(2):1255–1268
Bikash Rath Y, Rao G (2005) Bhetanoi-Balipadara Blackbuck Habitat. E-planet 9(2):42–49.
BirdLife International (2020) Important Bird Areas factsheet: Rollapadu Wildlife Sanctuary. Downloaded from http://www.birdlife.org.
Biswas S, Pawar SS (2006) Phylogenetic tests of distribution patterns in South Asia: towards an integrative approach. J Biosci 31:95–113
Clutton-Brock TH(1989) Review lecture: mammalian mating systems. Proceedings of the Royal Society of London. B. Biological Sciences, 236(1285), 339–372
Chen J, Li C, Yang J, Luo Z, Tang S, Li F, Li C, Liu B, Jiang Z (2012) Isolation and characterization of cross-amplification microsatellite panels for species of Procapra (Bovidae; Antilopinae). Int J Mol Sci 13(7):8805–8818
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014. https://doi.org/10.1093/oxfordjournals.jhered.a111627
Cornuet JM, Pudlo P, Veyssier J et al (2014) DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30:1187–1189. https://doi.org/10.1093/bioinformatics/btt763
Creel S, Spong G, Sands JL, Rotella J, Zeigle J, Joe L, Murphy KM, Smith D (2003) Population size estimation in yellowstone wolves with error-prone noninvasive microsatellite genotypes. Mol Ecol 12:2003–2009. https://doi.org/10.1046/j.1365-294X.2003.01868.x
De R, Kumar V, Ankit K, Khan KA, Kumar H, Kumar N, Goyal SP (2021) Cross-amplification of ungulate microsatellite markers in the endemic Indian antelope or blackbuck (Antilope cervicapra) for population monitoring and conservation genetics studies in south Asia. Mol Biol Rep 48(6):5151–5160
Dutta T, Sharma S, Maldonado JE et al (2013) Fine-scale population genetic structure in a wide-ranging carnivore, the leopard (Panthera pardus fusca) in central India. Divers Distrib 19:760–771. https://doi.org/10.1111/ddi.12024
Dutta T, Sharma S, Maldonado JE et al (2015) Genetic variation, structure, and geneflow in a sloth bear (Melursus ursinus) meta-population in the Satpura-Maikal landscape of central India. PLoS ONE 10:7–9. https://doi.org/10.1371/journal.pone.0123384
Eblate EM, Lughano KJ, Sebastian CD, Peter ML, Knut RH (2011) Polymorphic microsatellite markers for genetic studies of African antelope species. Afr J Biotechnol 10(56):11817–11820
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Foll M, Petit RJ (2009) Genetic consequences of range expansions. Annu Rev Ecol Evol Syst 40:481–501. https://doi.org/10.1146/annurev.ecolsys.39.110707.173414
Falconer DS (1996) Introduction to quantitative genetics. Pearson Education India, Noida
Frankel O, Soulé ME (1981) Conservation and evolution. Camb University Press, Cambridge
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10(6):1500–1508
Frankham R, Ballou SEJD, Briscoe DA, Ballou JD (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Futuyma DJ (1986) Evolutionary biology, 2nd edn. Sinauer, Sunderland, Mass
Fu Y (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Garcia-Cisneros A, Palacín C, Ben Khadra Y, Pérez-Portela R (2016) Low genetic diversity and recent demographic expansion in the red starfish Echinaster sepositus (Retzius 1816). Sci Rep 6:1–16. https://doi.org/10.1038/srep33269
Ghosh-Harihar M, An R, Athreya R et al (2019) Protected areas and biodiversity conservation in India. Biol Conserv 237:114–124. https://doi.org/10.1016/j.biocon.2019.06.024
Gilmour D (2018) The British in India: three centuries of ambition and experience. Penguin, UK
Goodman SJ, Tamate HB, Wilson R et al (2001) Bottlenecks, drift and differentiation : the population structure and demographic history of sika deer (Cervus nippon) in the Japanese archipelago. Mol Ecol 10(6):1357–1370
Greenwood PJ (1980) Mating systems, philopatry and dispersal in birds and mammals. Anim Behav 28:1140–1162. https://doi.org/10.1016/S0003-3472(80)80103-5
Gürler Ş, & Bozkaya F. (2013). Genetic diversity of three native goat populations raised in the South-Eastern region of Turkey. Kafkas Üniversitesi Veteriner Fakültesi Dergisi 19(2), 207-213
Hora SL (1949) Satpura hypothesis of the distribution of the Malayan fauna and flora to peninsular India. Proc Natl Inst Sci India 15:309–314
Horváth MB, Martínez-Cruz B, Negro JJ, Kalmár L, Godoy JA (2005) An overlooked DNA source for non‐invasive genetic analysis in birds. J Avian Biol 36(1):84–88
Hughes JE (2009) Animal kingdoms: princely power, the environment, and the hunt in colonial India. UT Austin Diss
Hume BCC, Voolstra CR, Arif C et al (2016) Ancestral genetic diversity associated with the rapid spread of stress-tolerant coral symbionts in response to Holocene climate change. PNAS 113:4416–4421. https://doi.org/10.1073/pnas.1601910113
IUCN SSC Antelope Specialist Group (2017) The IUCN Red List of Threatened species 2017: e.T1681A50181949
Jana A, Karanth P (2019) Multilocus nuclear markers provide new insights into the origin and evolution of the blackbuck (Antilope cervicapra, Bovidae). Mol Phylogenet Evol 139:106560. https://doi.org/10.1016/j.ympev.2019.106560
Jhala YV, Giles RH (1991) The status and conservation of the wolf in Gujarat and Rajasthan, India. Conserv Biol 5:476–483. https://doi.org/10.1111/j.1523-1739.1991.tb00354.x
Joshi A, Vaidyanathan S, Mondo S et al (2013) Connectivity of tiger (Panthera tigris) populations in the human-influenced forest mosaic of central India. PLoS ONE. https://doi.org/10.1371/journal.pone.0077980
Kearse M, Moir R, Wilson A et al (2012) Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. https://doi.org/10.1093/bioinformatics/bts199
Kerth G, Mayer F, König B (2000) Mitochondrial DNA (mtDNA) reveals that female Bechstein’s bats live in closed societies. Mol Ecol 9:793–800. https://doi.org/10.1046/j.1365-294X.2000.00934.x
Knowles LL (2000) Tests of pleistocene speciation in montane grasshoppers (Genus melanoplus) from the sky islands of western North America. Evolution (N Y) 54:1337–1348. https://doi.org/10.1111/j.0014-3820.2000.tb00566.x
Kumar V, Sharma N, Singal K, Sharma A (2018) A wildlife forensic study for the species identification of Indian Blackbuck through forensically informative nucleotide sequencing (FINS). Biosci Biotechnol Res Asia 15(1):175
Kurup GU (1974) Mammals of Assam and the mammal-geography of India. In: Mani MS (ed) Ecology and biogeography in India. Dr. W. Junk b.v, Publishers, The Hague, pp 585–613
Lappan S (2007) Patterns of dispersal in Sumatran Siamangs (Symphalangus syndactylus): preliminary mtDNA evidence suggests more frequent male than female dispersal to adjacent groups. Am J Primatol. https://doi.org/10.1002/ajp.20382
Lukoschek V (2018) Population declines, genetic bottlenecks and potential hybridization in sea snakes on Australia’s Timor Sea reefs. Biol Conserv 225:66–79. https://doi.org/10.1016/j.biocon.2018.06.018
Mahmoudi B, Babayev MS (2009). The investigation of genetic variation in Taleshi goat using microsatellite markers. Res J Biol Sci 4(6): 644-646
Mallon DP (2008) Antilope cervicapra. The IUCN red list of threatened species 2008: e.T1681A6448761. https://doi.org/10.2305/IUCN.UK.2008.RLTS.T1681A6448761.en
Mallon DP, Kingswood SC(2001) (compilers). Antelopes. Part 4: North Africa, the Middle East, and Asia. Global Survey and Regional Action Plans. SSC Antelope Specialist Group. IUCN, Gland, Switzerland and Cambridge, UK. viii + 260pp
Mamatha MD, Hosetti BB (2018) Census study on blackbucks Antilope cervicapra (L.) in Hullathi section of Ranebennur Wildlife Sanctuary (RWLS), Ranebennur, Haveri district, Karnataka. Int J Zool Appl Biosci 3:283–288
Mani MS (1974) Physical features. In: Mani MS (ed) Ecology and Biogeography in India. Dr. W. Junk b.v Publishers, The Hague, pp 11–58
Maudet, F., Maillard, J. C., Chardonnet, P.,& Sauzier, J. (1999). Study of genetic diversity in a Rusa deer (Cervus timorensis russa) population in Mauritius using derived bovine microsatellites. In Third Annual Meeting of Agricultural Scientists (p.131)
Meena R, Saran RP (2018) Distribution, ecology and conservation status of blackbuck (Antilope cervicapra): an update. Int J Biol Res 3:79–86
Modolo L, Salzburger W, Martin RD (2005) Phylogeography of Barbary macaques (Macaca sylvanus) and the origin of the Gibraltar colony. PNAS 102:7392–7397
Movahedin, M. R., Amirinia, C., Noshary, A., & Mirhadi, S. A. (2010) Detection of genetic variation in sample of Iranian proofed Holstein cattle by using microsatellite marker. Afr J Biotechnol 9(53): 9042-9045
Moore WS (1995) Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution (N Y) 49:718–726. https://doi.org/10.2307/2411136
Morin PA, Chambers KE, Boesch C, Vigilant L (2001) Quantitative polymerase chain reaction analysis of DNA from noninvasive samples for accurate microsatellite genotyping of wild chimpanzees (Pan troglodytes verus). Mol Ecol 10(7):1835–1844
Mukherjee S, Krishnan A, Tamma K et al (2010) Ecology driving genetic variation: a comparative phylogeography of jungle cat (Felis chaus) and leopard cat (Prionailurus bengalensis) in India. PLoS ONE. https://doi.org/10.1371/journal.pone.0013724
Mungall EC(1978) The Indian blackbuck antelope: a Texas view (No. QL737. M86 1978.)
Nag KSC, Pramod P, Karanth KP (2011) Taxonomic implications of a field study of morphotypes of hanuman langurs (Semnopithecus entellus) in Peninsular India. Int J Primatol 32:830–848. https://doi.org/10.1007/s10764-011-9504-0
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Noss RF (1990) Indicators for monitoring biodiversity: a hierarchical approach. Conserv Biol 4:355–364
Nutt KJ (2008) A comparison of techniques for assessing dispersal behaviour in gundis: Revealing dispersal patterns in the absence of observed dispersal behaviour. Mol Ecol 17:3541–3556. https://doi.org/10.1111/j.1365-294X.2008.03858.x
Orrú L, Napolitano F, Catillo G, Moioli B (2006) Meat molecular traceability: How to choose the best set of microsatellites?. Meat Sci 72(2):312–317
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Peakall R, Smouse PE (2012) GenALEx 6.5: genetic analysis in excel. Population genetic software for teaching and research-an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Polzin T, Daneshmand SV (2001) Improved algorithms for the Steiner problem in networks. Discret Appl Math 112:263–300. https://doi.org/10.1016/S0166-218X(00)00319-X
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nat Rev Genet 6(11):847–859
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Ramachandran V, Robin VV, Tamma K, Ramakrishnan U (2017) Climatic and geographic barriers drive distributional patterns of bird phenotypes within peninsular India. J Avian Biol 48:620–630. https://doi.org/10.1111/jav.01278
Ranjitsinh MK (1989) The Indian blackbuck. Natraj Publishers, Dehradun
Rasmussen AL, Okumura A, Ferris MT et al (2014) Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Sci (80-) 346:987–992
Reddy S (2014) What’s missing from avian global diversification analyses? Mol Phylogenet Evol 77:159–165. https://doi.org/10.1016/j.ympev.2014.04.023
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237. https://doi.org/10.1046/j.1523-1739.2003.01236.x
Ripley SD, Beehler BM (1990) Patterns of speciation in Indian birds. J Biogeogr 17:639–648
Rivers MC, Brummitt NA, Lughadha E, Meagher TR (2014) Do species conservation assessments capture genetic diversity ? Glob Ecol Conserv 2:81–87. https://doi.org/10.1016/j.gecco.2014.08.005
Røed, K. H. (1998). Microsatellite variation in Scandinavian cervidae using primers derived from Bovidae. Hereditas, 129(1), 19-25
Roonwal ML (1984) Tail form and carriage in Asian and other primates, and their behavioral and evolutionary significance. In: Roonwal ML, Mohnot SM, Rathore NS (eds) Current primate research. Jodhpur University Press, Jodhpur, India. 93–151
Roth, T., Pfeiffer, I., Weising, K., & Brenig, B. (2006). Application of bovine microsatellite markers for genetic diversity analysis of European bison (Bison bonasus). J Anim Breed Genet 123(6): 406-409
Roy S, Sen (2019) Spatial patterns of trends in seasonal extreme temperatures in India during 1980–2010. Weather Clim Extrem 24:100203. https://doi.org/10.1016/j.wace.2019.100203
Rozas J, Ferrer-Mata A, Sanchez-DelBarrio JC et al (2017) DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol 34:3299–3302. https://doi.org/10.1093/molbev/msx248
Sabuni CA, Van Houtte N, Gryseels S et al (2016) Genetic structure and diversity of the black and rufous sengi in Tanzanian coastal forests. J Zool 300:305–313. https://doi.org/10.1111/jzo.12384
Schaller GB (2009) The deer and the tiger. University of Chicago Press, Chicago
Sharma DK, Maldonado JE, Jhala YV, Fleischer RC (2004) Ancient wolf lineages in India. Proc R Soc B Biol Sci 271:2–5. https://doi.org/10.1098/rsbl.2003.0071
Slate J, Coltman DW, Goodman SJ, MacLean I, Pemberton JM, Williams JL (1998) Bovine microsatellite loci are highly conserved in red deer (Cervus elaphus), sika deer (Cervus nippon) and Soay sheep (Ovis aries). Anim Genet 29(4):307–315
Sonsthagen SA, Wilson RE, Underwood JG (2017) Genetic implications of bottleneck effects of differing severities on genetic diversity in naturally recovering populations: an example from Hawaiian coot and Hawaiian gallinule. Ecol Evol 7:9925–9934. https://doi.org/10.1002/ece3.3530
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24:3189–3194. https://doi.org/10.1093/nar/24.16.3189
Tajima F (1996) The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 1465:1457–1465
Tamma K, Ramakrishnan U (2015) Higher speciation and lower extinction rates influence mammal diversity gradients in Asia. BMC Evol Biol 15:1–13. https://doi.org/10.1186/s12862-015-0289-1
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Thangaraj K, Prathap Naidu B, Crivellaro F et al (2010) The influence of natural barriers in shaping the genetic structure of Maharashtra populations. PLoS ONE 5:1–9. https://doi.org/10.1371/journal.pone.0015283
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Vial L, Maudet C, Luikart G (2003) Thirty‐four polymorphic microsatellites for European roe deer. Mol Ecol Notes 3(4):523–527
Vidya TNC, Fernando P, Melnick DJ, Sukumar R (2005a) Population differentiation within and among Asian elephant (Elephas maximus) populations in southern India. Heredity (Edinb) 94:71–80. https://doi.org/10.1038/sj.hdy.6800568
Vidya TNC, Fernando P, Melnick DJ, Sukumar R (2005b) Population genetic structure and conservation of Asian elephants (Elephas maximus) across India. Anim Conserv 8:377–388. https://doi.org/10.1017/S1367943005002428
Vidya TNC, Sukumar R, Melnick DJ (2009) Range-wide mtDNA phylogeography yields insights into the origins of Asian elephants. Proc R Soc B Biol Sci 276:893–902. https://doi.org/10.1098/rspb.2008.1494
Wang GZ, Chen SS, Chao TL, Ji ZB, Hou L, Qin ZJ, Wang JM (2017) Analysis of genetic diversity of Chinese dairy goats via microsatellite markers. J Anim Sci 95(5):2304–2313
Wang W, Zheng Y, Zhao J, Yao M (2019) Low genetic diversity in a critically endangered primate: shallow evolutionary history or recent population bottleneck? BMC Evol Biol 19:1–13. https://doi.org/10.1186/s12862-019-1451-y
Waterman J (2008) Male mating strategies in rodents Rodent societies. University of Chicago Press, Chicago, pp 27–41
Wereszczuk A, Leblois R, Zalewski A (2017) Genetic diversity and structure related to expansion history and habitat isolation: Stone marten populating rural-urban habitats. BMC Ecol 17:1–16. https://doi.org/10.1186/s12898-017-0156-6
White OL, Densmore LD III (1992) Mitochondrial DNA isolation. Molecular genetic analysis of populations, a practical approach, 1st edn. IRL Press, Oxford, pp 29–55
Willis SC, Nunes M, Montaña CG et al (2010) The Casiquiare river acts as a corridor between the Amazonas and Orinoco river basins: Biogeographic analysis of the genus Cichla. Mol Ecol 19:1014–1030. https://doi.org/10.1111/j.1365-294X.2010.04540.x
Worley K, Strobeck C, Arthur S et al (2004) Population genetic structure of North American thinhorn sheep (Ovis dalli). Mol Ecol 13:2545–2556. https://doi.org/10.1111/j.1365-294X.2004.02248.x
Funding
This work was supported by grants to PK under the partnership between the Department of Biotechnology, Govt. of India, and Indian Institute of Science (DBT-IISc partnership) and permits to AJ from the state forest departments of Karnataka, Andhra Pradesh, Tamil Nadu, Maharashtra and Gujarat and the village panchayat of Bhetanai in Orissa. AJ would like to thank Bibidishananda Basu and Kavya Lakshmikanth for helping out with part of the wet-lab work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Ethical approval
All sampling was non-invasive, and the animals were not stressed or harmed in any manner.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jana, A., Karanth, K.P. Not all is black and white: phylogeography and population genetics of the endemic blackbuck (Antilope cervicapra). Conserv Genet 24, 41–57 (2023). https://doi.org/10.1007/s10592-022-01479-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-022-01479-x