
Abstract
Conserving biodiversity in an era of rapid climate change requires understanding the mechanisms that influence dispersal, gene flow and, ultimately, species persistence. This information is becoming critical for conserving key species in rapidly warming places such as the Arctic. Arctic freshwater fish not only face warmer conditions, but also the drying of tundra streams due to climate change. Here, we examined population structure, gene flow, and the influence of landscape features on the neutral genetic variation of the Arctic grayling on Alaska’s North Slope. We estimated the number of genetically distinct clusters and determined effective population sizes for and patterns of gene flow among geographic regions. We predicted that river distance, river drying, distance to the coast, and elevational gradient would influence genetic differentiation for Arctic grayling. Bayesian clustering and discriminant analysis of principal components found support for five or six genetic clusters roughly corresponding to downstream and headwater subwatersheds. Estimates of gene flow revealed asymmetric downstream bias. River distance and river dry zones were significantly associated with increasing genetic differentiation among sampling locations despite this species' high dispersal capability and the temporary nature of dry zones. Isolation and downstream-biased dispersal could contribute to high levels of inter-population genetic variation among the headwaters of the North Slope Arctic grayling metapopulation, which might be particularly important for species conservation during rapid climate change. More generally, small, isolated populations might drive particular alleles to higher frequencies due to selection or drift, thus promoting the genetic potential for rapid evolutionary changes under future climate change.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
To persist through this era of rapid climate change, species must either adjust to new conditions through evolution, employ phenotypic plasticity, or track suitable habitat through dispersal or gene flow. Gaps in understanding these responses reduces our ability to predict the fate of individual populations, species, and natural ecosystems (Parmesan 2006; Scheffers et al. 2016; Urban et al. 2016). In particular, estimating dispersal across natural landscapes often requires quantifying the effect of temporary, but important, barriers that appear during critical periods of movement (Manel et al. 2003; Richardson 2012). These landscape barriers to movement not only can affect demographic dynamics, but also gene flow, the transfer of genes among populations, which in turn can influence population persistence by altering genetic diversity and adaptability (Hoffmann and Sgrò 2011; Schloss et al. 2012; Urban et al. 2013). Thus, population genetics is especially useful for discovering cryptic barriers to movement and assessing their effect on genetic variation across natural landscapes.
Freshwater river species are particularly sensitive to habitat fragmentation and genetic isolation due to the limited number of movement pathways defined by their dendritic river systems. Population isolation due to river fragmentation can decrease freshwater species genetic variation and increase extinction risk (Fagan 2002). Anthropogenic barriers, such as dams and culverts, often restrict the dispersal of aquatic organisms (Junker et al. 2012; Peterson and Ardren 2009; Roberts et al. 2013). Natural environmental drivers, such as droughts and floods (Fitzpatrick et al. 2014; Hopken et al. 2013; Meeuwig et al. 2010; Perkin et al. 2015) or changes to the location of spawning areas and overwintering refugia (Fausch et al. 2002; Kanno et al. 2011; Maria et al. 2012; Ozerov et al. 2012; Vähä et al. 2007), also influence dispersal, genetic structure, and extinction risk for numerous freshwater species (Fagan et al. 2007; Kanno et al. 2011; Mossop et al. 2015; Poissant et al. 2005). Many freshwater species display elevated levels of genetic isolation compared to other geographically structured populations (Morrissey and Kerckhove 2009), possibly due to semi-isolated upstream populations with downstream-biased dispersal. Effective conservation requires a better understanding of how changing environments influence population structure, genetic differentiation, and extirpation (Campbell Grant et al. 2007; Fagan 2002; Labonne et al. 2008).
Arctic grayling (Thymallus arcticus) play a keystone role in the Arctic by integrating stream and lake ecosystems through their annual migrations between the two systems. Clear, cool, low-order streams provide spring spawning and summer feeding and rearing habitats for Arctic grayling. However, because most tundra streams freeze solid in winter, current evidence indicates that grayling either must overwinter in deep lakes, higher-order coastal streams, or other locations that resist freezing (Beauchamp 1990; Parkinson et al. 1999; West et al. 1992). Appropriate spawning, feeding, and overwintering habitats can occur more than 100 km apart, thus necessitating extensive long-distance seasonal migrations among habitats (West et al. 1992). As the climate warms, sections of Tundra rivers are becoming dry and impassable when summer evapotranspiration exceeds precipitation, causing breaks in aquatic connectivity (ACIA 2005; Hinzman et al. 2005; Martin et al. 2009). This increased frequency and duration of river drying could restrict fish movement (Betts and Kane 2015; Junker et al. 2012; Primmer et al. 2006; Reist et al. 2006), potentially altering dispersal and gene flow. Grayling are restricted to freshwater habitats and rarely occur in near-shore habitats, unlike other salmonids which can tolerate higher salinities (Northcote 1995; Blair et al. 2016). Due to their reliance on a variable freshwater network, Arctic grayling population persistence likely depends on the spatial and temporal connectivity of this freshwater environment (Opdam and Wascher 2004). However, no studies have identified what factors might limit dispersal and gene flow or if climate-induced fragmentation might alter population structure or persistence for this keystone fish.
Here, we applied population genetics to assess the number of potential genetic clusters, determine effective population size, estimate gene flow, and identify the landscape features that influence fragmentation and genetic differentiation for the freshwater salmonid, Arctic grayling, on Alaska’s North Slope, a region undergoing the fastest rates of climate change (IPCC 2013). We predicted that landscape features would strongly influence genetic differentiation among sampled locations and geographic regions as indicated by patterns of isolation by distance and isolation by environment (Table 1; Meffe and Vrijenhoek 1988; Stamford and Taylor 2004; Lowe et al. 2006; Jenkins et al. 2010; Perkin et al. 2015). Alternatively, the Arctic grayling’s long-distance dispersal ability might override the effects of isolation by distance and environment, thereby fostering a panmictic distribution and potentially offering simpler decisions regarding its management and conservation.
Methods
Site description
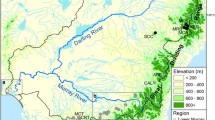
We worked in the Itkillik, Kuparuk, and Sagavanirktok watersheds that drain the foothills of the Brooks Range (Fig. 1, Site Map inset). The Kuparuk and Sagavanirktok rivers drain directly into the Arctic Ocean, whereas the Itkillik drains into the Colville River ~ 40 km from the Arctic Ocean. These watersheds formed during the middle to late Pleistocene (Hamilton 2003), originate within the Brooks Mountains, drain northward to the Arctic Ocean, and are separated at their mouths by coastal estuarine habitat. We chose these three watersheds because of their observed differences in their susceptibility to river drying: the Sagavanirktok upper watershed (including Oksrukuyik Creek) harbors many potential dry zones along its length, the Kuparuk watershed is less susceptible to dry zones, but still contains temporary dry zones, and the Itkillik watershed is well-connected along its length to the coast. We chose 15 sampling locations to stratify habitats within and among these watersheds and to represent locations separated by specific dry river zones (Fig. 1; Table S1). Additionally, we incorporated the Ublutuoch River on the coastal plain located approximately 25 km from the Arctic Ocean, close to the mouth of the Colville River and its connections to the upstream Itkillik reach. Hereafter, we use the term, geographic region, to group sampling locations into a set of pre-defined locations, corresponding to hypothesized genetic differentiation (1) among the three watersheds and (2) between upstream (near headwater lakes) and downstream locations (in larger rivers that drain to the coast) that could be separated by river dry zones.

Map of study area and (A–C) and Arctic grayling population structure (D). On the map at left, we display the overall study region (A) on Alaska’s North Slope (C), including a finer scaled inset map (B) of sampling locations in the foothills of the Brooks Range. The Colville/Itkillik, Kuparuk, and Sagavanirktok watersheds flow from the headwaters in the Brooks Range north to the Arctic Ocean. At each sampling location, different symbols depict different watersheds, and colors correspond to the most dominant genetic cluster at that location. The gray color for K7 indicates a mixture of all genetic clusters. To the right (D), STRUCTURE plots depict individual assignment probabilities to six genetic clusters (indicated by color) for individuals captured at each of the 16 sampling locations. Freshwater stream sections are show in blue, coastal sections are shown in green, and intermittently dry sections are shown in red. Thick red lines indicate dry zones > 1.5 km in extent, and narrow red lines indicate dry zones < 1.5 km in extent. Number of samples are indicated above each subplot. Full site names and characteristics can be found in Table S1
Fish sampling
We sampled adult Arctic grayling from May to August from 2010 to 2013 after fish had migrated into the streams from their overwintering locations. We collected approximately 30 individuals from each sampling location to determine population genetic structure and assess genetic differentiation among locations. Due to logistically challenging conditions on the North Slope, we only collected 22 individuals from the Lower Sagavanirktok River and nine individuals from the Lower Kuparuk River. Here we use the term, lower, to indicate regions downstream from upstream regions of each watershed rather than anything regarding stream gradient. We captured Arctic grayling using a combination of seine, fyke, and gill nets, as well as via hook and line. We collected caudal fin tissue from each individual, preserved the tissue in 95% ethanol and stored the preserved samples at −20 °C until DNA extractions were conducted.
Microsatellites and descriptive statistics
DNA was extracted from fin tissue using DNeasy blood and tissue kits (Qiagen, CA). Multiplex polymerase chain reactions (PCRs) were optimized for allelic range for twelve highly variable nuclear microsatellite markers specific to Arctic grayling (Diggs and Ardren 2008) following Steed (2007) (Table S2). PCR products were analyzed on an ABI 3130xl DNA sequencer with GeneScan 500(−250) LIZ size standard. Allele sizes were scored along with positive and negative controls using the program GeneMarker (Softgenetics, LLC, State College, PA). Output genotypes with amplifications that were too weak to resolve peaks or had excess stutter were re-amplified and rerun for better resolution and to ensure accuracy. Any remaining unresolved alleles were treated as missing data. We ran multiple individuals as positive controls, with at least one positive control per well plate, and using multiple positive controls to ensure consistent peaks were obtained from run to run. In the few cases where we received different results for controls, we re-ran the samples.
We screened for null alleles, large allele dropout, and scoring errors using the program MICRO-CHECKER v.2.2.3 (Van Oosterhout et al. 2004). Exact tests (Guo and Thompson 1992) were used to test for deviations from Hardy–Weinberg equilibrium across all loci and all regions with 1,000,000 Markov Chain Monte Carlo (MCMC) and 100,000 dememorization steps in the program ARLEQUIN v3.5 (Excoffier and Lischer 2010). We also used ARLEQUIN v3.5 to test for deviations from linkage equilibrium across all pairs of loci using an expectation–maximization algorithm with 10,000 permutations. Probability values were Bonferroni-corrected whenever multiple testing occurred. We calculated descriptive statistics for allelic richness and private alleles using the program GenoDive 2.0b23 (Meirmans and Van Tienderen 2004) and observed and expected heterozygosity using ARLEQUIN v3.5. Unbiased estimates of allelic richness and private alleles per sampling location were calculated via rarefaction using the program HP-Rare 1.0 (Kalinowski 2005).
Analysis of molecular variance (AMOVA)
We used analysis of molecular variance (AMOVA) in the program GenoDive 2.0b23 with both an infinite alleles model (F-statistics) and a stepping-stone model (R-statistics) with 10,000 iterations to investigate partitioning of genetic variation within and among sampled locations. We first partitioned variance among individuals (FIT), among individuals within sampled locations (FIS) and among sampled locations (FST). We also conducted a hierarchical AMOVA in R using the poppr package (Kamyar et al. 2014) to provide assessment of broad-scale regional distribution of genetic variation. Individual genotypic data were first converted to Nei’s genetic distances and then used in a hierarchical model to investigate nested regional population structure. We partitioned genetic variation among individuals within regions defined by the three major watersheds (FRT) and within and among sub-regions defined by sub-watersheds (FSR and FST, respectively). We used a more detailed approach to investigate the correlation of landscape features with genetic differentiation (see Sect. 2.3 Landscape Genetics). Besides generating broad-scale F-statistics, we determined pairwise genetic differentiation (pairwise FST) and its significance among sampling locations using ARLEQUIN v3.5 and GENEPOP (Rousset 2008) with the Markov Chain parameterized using 10,000 dememorization steps, 100 batches and 5000 iterations per batch. We compared values of pairwise FST with pairwise G’ST and Jost’s D and found that all metrics produced similar results (Table S3). Ultimately, we chose to use pairwise F-statistics throughout this manuscript for their appropriateness for assessing deviations from panmixia and analyzing potential barriers to gene flow (Whitlock 2011).
Genetic structure
We conducted analyses to estimate the number of genetically determined clusters represented by our data. Genetic structure was inferred using two complementary approaches: Bayesian assignment in STRUCTURE (Pritchard et al. 2000) and discriminant analysis of principal components (DAPC) within the adegenet package (Jombart et al. 2010) in R. STRUCTURE was used to estimate the number of genetic clusters (K) using the log likelihood of individual assignment into a range of potential genetic clusters. We used the genetic admixture option with a burn-in length of 25,000 iterations preceding each MCMC simulation (100,000 iterations for K = 1 to 12, repeated 20 times for each value of K). We used CLUMPP (Jakobsson and Rosenberg 2007) to combine results of separate STRUCTURE runs and DISTRUCT (Rosenberg 2003) to visualize solutions for different genetic clusters. STRUCTURE HARVESTER (Earl and VonHoldt 2011) was used to assess and visualize likelihood values, including ΔK, which evaluates the net rate of change moving from one K to the next (Evanno et al. 2005). We used CLUMPAK (Li and Liu 2018) to calculate the MedMeaK, MaxMeaK, MedMedK and MaxMedK estimates of number of genetic clusters, which are less susceptible to biases introduced by uneven sampling (Puechmaille 2016).
We used multiple methods to evaluate support for various numbers of genetic clusters (K) in our data, including STRUCTURE log probability, the ΔK method, BIC from a DAPC analysis, MedMeaK, MaxMeaK, MedMedK and MaxMedK, as well as statistical similarities and differences in pairwise FST among sampling locations (Table 2, S4–S5). DAPC produced Bayesian Information Criterion (BIC) values indicating support for K = 3–5 genetic clusters (Fig. S1B). Based on support for larger numbers of genetic clusters in STRUCTURE, we chose the highest value of K = 5 for the DAPC analysis. The log probability of data for STRUCTURE analyses peaked at K = 5 and 7 (Supplementary Fig. S1A). For ΔK, we found peaks at K = 2, 5 and 7 (Supplementary Fig. S2). However, MedMeaK, MaxMeaK, MedMedK and MaxMedK indicated K values from 6–7. Based on these inferences, we chose a compromise value of K = 6 genetic clusters for STRUCTURE, which includes the value suggested by the less biased MedMeaK/MedMedK approach, and is close to the K = 5 supported by the DAPC approach. However, choosing alternative cluster numbers from K = 3 to K = 7 did not qualitatively change the overall interpretation of our results (Supplementary Fig. S3).
Effective population size
We measured effective population size (Ne) via the linkage-disequilibrium method of Nomura (2008). We used the program NeEstimator V2.1 (Do et al. 2014) to evaluate broad-scale regional differences in Ne (England et al. 2006). We estimated Ne for geographic regions within each watershed based on location (upstream or downstream) to evaluate if effective population size was greater in downstream regions due to downstream flow and upstream barriers (Table 1) and between areas within watersheds separated by significant dry zones. Downstream locations included S7 within the Sagavanirktok watershed, K7 within the Kuparuk watershed, and C2, the Ublutuoch, close to the mouth of the main Colville River (Fig. 1; Table S1). Other regions consisted of sampling locations within sub-watersheds located near headwater areas, including the Upper Sagavanirktok (S1 to S4) versus the Oksrukuyik (S5 and S6) separated by a substantial dry zone, the Upper Kuparuk (K1 to K4) versus Toolik (K5 and K6) separated by a substantial dry zone, and Upper Itkillik/Colville (C1), which is far upstream from C2, but is likely not interrupted by dry zones. Here we define substantial dry zones as those stream reaches with no surface expression of water for more than 7 days during the Arctic open-water period from early May to mid-September. Location and number of river dry zones were defined using GPS coordinates from helicopter flight surveys and ground surveys as well as with game cameras and/or temperature and pressure loggers placed at various locations throughout our study system. We chose consensus values of Ne for each geographic region based on allele frequencies using Pcrit, defined as 1/(2S) where S is the sample size (Do et al. 2014). Similar to rarefication, this method reduces the downward bias that occurs when sample size is low and the potential to miss rare alleles is high and reduces upward bias when sample size is relatively high (England et al. 2006; Waples and Do 2010). Because single-sample Ne estimates tend to be biased downward for iteroparous species due to potential mixed age-class Wahlund effect (Waples et al. 2014), we interpreted our estimates as relative, not absolute, estimates for effective population size comparison and dispersal potential among regions.
Rates of gene flow
We estimated contemporary bi-directional rates of gene flow over the last two generations using BAYESASS v3.0 (Wilson and Rannala 2003). Because genetic migration terminology can be confusing when discussing migratory species, we use the term migration to refer to seasonal movement of individuals between habitats without genetic displacement from local populations, gene flow to refer to movement rate of genes from one population to another, and dispersers to refer to individuals contributing genes via dispersal from one population to another. We used the same geographic regions discussed for Ne to test hypotheses regarding gene flow within and among watersheds. BAYESASS3 is a Bayesian resampling method that provides estimates of recent asymmetrical gene flow by determining the proportion of individuals in each region with non-resident ancestry. Recent dispersers and their progeny display genotypic disequilibrium relative to the population from which they were sampled (Wilson and Rannala 2003). The program assumes that background gene flow is relatively low (FST > 0.05) and that loci are in linkage equilibrium, but it allows for deviations from Hardy–Weinberg equilibrium by estimating population-specific inbreeding coefficients (Faubet et al. 2007). We obtained estimates of posterior mean gene flow and the standard deviation of the marginal posterior distribution using a random starting seed, 20,000,000 MCMC iterations, a burn-in of 3,000,000 iterations, and a sampling interval every 100 iterations. We adjusted the dispersal, allele frequency, and inbreeding coefficient mixing parameters to 0.1, 0.2, and 0.2, respectively, which ensured that the proposed changes between chains were between 40 and 60% of the total number of iterations (Rannala 2007). BAYESASS3 estimates of recent gene flow can vary in accuracy depending on adherence to model assumptions and degree of genetic differentiation among populations (Faubert et al. 2007). We assessed accuracy of our gene flow estimates based on precision, convergence, and deviance of multiple model runs (Meirmans 2014) by examining trace files from multiple BAYESASS3 runs that differed in starting seed using the program TRACER v1.7 (Rambaut et al. 2003) and by comparing model deviance (Meirmans 2014).
Landscape genetics
Landscape genetic analyses investigate correlations between the genetic differentiation of individuals sampled across geographic locations and corresponding landscape features. Hierarchical AMOVA, discussed above, is limited in its ability to evaluate factors associated with genetic differentiation by the way it partitions genetic variation broadly among nested geographic areas. We used more detailed landscape genetics models to investigate potential correlations with environmental features using redundancy analysis (RDA) and Moran’s eigenvector maps (MEMs). These landscape genetics models use spatial and ecological predictor variables measured among sampled locations to explain observed differences in genetic variation (Manel and Helderegger 2013; Storfer et al. 2010). We assessed a priori predictions listed in Table 1 about the influence of river distance (km), river dry zone distance (km), estuary distance (km), and elevation (m) on genetic variation among individuals sampled at 16 locations.
Environmental matrices were derived using the STARS ArcGIS toolset (Peterson and Ver Hoef 2014) in ARCMAP v10.2 (ESRI 2013). GIS data included a digital elevation model (SDMI 2013) and stream and water body shapefiles (USGS 2014). River dry zones were identified in the field during helicopter and ground surveys, monitored with game cameras and/or temperature and pressure loggers, and mapped using GPS coordinates during the Arctic open-water period from early May to mid-September 2010 to 2014 (Fig. 1, red lines). Our genetic distance matrix consisted of pairwise standardized FST obtained using individual genotype data in the program GenoDive 2.0b23, described above. Matrices for genetic distance, river distance, river dry zone distance, and other environmental covariates are included in Tables S4 and S5.
We removed spatial dependencies within our distance matrices using Moran’s eigenvector maps (MEMs), a more powerful approach than the alternative Mantel test. Using the R package memgene (Galpern et al. 2014), we converted pairwise environmental variables into sets of site-based environmental MEM vectors (Table S5). Then for each set of MEM vectors, we used forward selection redundancy analysis (RDA) against the pairwise FST matrix to identify variables significantly explaining genetic variation among locations (significance level P < 0.05 after 10,000 random permutations). RDA combines multivariate multiple linear regression analysis with principal components analysis to model multivariate genetic response data. Significant MEM vectors were combined to create a set of explanatory variables used in a final RDA in the R package vegan (Oksanen et al. 2013). In addition to the environmental variables described here, we tested alternative models using similar collinear environmental factors, including number of river dry zones instead of dry zone distance and watershed association, where sample locations are either from the same or different watersheds, instead of coastal distance. We obtained similar model results when we substituted factor variables for distance variables (Table S6A), and chose to present our analysis based on environmental distances. Results obtained using Mantel tests and partial Mantel tests with the same environmental variables produced similar results to those obtained using RDA with MEM transformed variables (Table S6B).
Results
Microsatellite screening and descriptive statistics
Of the 12 original microsatellite loci, two loci (Tar109 and Tar112) exhibited homozygote excess in five of 16 sampling locations and evidence of either null alleles, large allele dropout, or scoring errors (Van Oosterhout et al. 2004). Although variation from population genetics null models might reflect multiple subpopulations within sampled locations (Wahlund 1928; Nei and Li 1973), these two loci were consistently difficult to score and were removed from further analyses as a precautionary measure due to the high likelihood of scoring errors. Our final dataset included 10 loci and a total of 437 individuals from 16 sampling locations. Across the remaining 10 loci, we found no significant deviations from Hardy Weinberg equilibrium, except for a single locus (Tar114) in one sampling location (Oksrukuyik Creek) with P < 0.0003 (Table S7 and Fig. S4). We found no evidence of linked loci. All remaining loci were highly polymorphic with a mean of 32.2 ± 6.3 alleles per locus (Table S8) and location-specific gene diversity ranging from 0.84 to 0.93 (Table S9). Number of alleles per sampling location varied from 11 to 20 alleles with similar patterns of diversity reflected in effective number of alleles, rarified allelic richness, rarified private allele richness and heterozygosity (Table S1). Within the Sagavanirktok and Kuparuk watersheds, private allele richness increased from 0.2 for furthest upstream headwater locations (S1, S5, K1, and K5) to 0.4 for downstream-most locations (S7 and K7), with similar patterns of increasing diversity from headwaters to downstream for all indices (Table S1). The two locations within the Colville/Itkillik watershed (C1 and C2) had the highest allelic richness compared to all other locations. In a post hoc analysis, we found a pattern of increasing heterozygosity with increasing latitude (r = 0.176, P = 0.026) using a linear model in R, but no relationship after removing our only high latitude location, the Ublutuoch (C2) on the coastal plain, from the analysis (Fig. S5).
Broad-scale patterns of genetic differentiation
Hierarchical nested analysis of molecular variance (AMOVA) indicated lower than expected genetic variance within sub-watersheds, higher than expected genetic variance among sub-watersheds, and higher than expected genetic variance among watersheds (P value = 0.0001 for all levels, Fig. S6). The highest percentage of covariance (91%) occurred among sub-watersheds (FST = 0.088, p < 0.001). AMOVA based on sampling locations indicated that most of the genetic variation occurred among individuals within the total population (FIT = 0.057, RIT = 0.088) and 4–7% of genetic variation could be accounted for by genetic differences among sampling locations (FST = 0.041, RST = 0.065), which underscores the importance of regional geographic associations in this system. These broad-scale AMOVA results support results of regional population structure found in STRUCTURE and DAPC analyses. Patterns of pairwise FST among sampling locations also supported these patterns of genetic differentiation across our study area. Clusters of sampling locations with low pairwise FST corresponded to distinct geographic regions (Table 2). Sampling locations with low among-location genetic differentiation included the following groups: S1, S2, S3, S4, and S7 (FST ≤ 0.01); S5 and S6 (FST ≤ 0.01); K1, K2, K3, K4, and K7 (FST ≤ 0.01); K5 and K6 (FST ≤ 0.002); and C1 and C2 (FST ≤ 0.016).
Genetic structure
Results from STRUCTURE and DAPC further indicated significant and consistent genetic structure across our study area (Figs. 1, 2). Assignment probability to genetic clusters varied among individuals with specific genetic clusters dominating distinct geographic regions (Fig. 1). STRUCTURE analysis indicated that cluster 1 (green) dominated the Colville/Itkillik watershed (C1–2), cluster 2 (blue) dominated the upper Kuparuk watershed (K1–4), and cluster 3 (purple) dominated the Toolik tributary of the Kuparuk (K5–6), upstream from a substantial dry zone. The Mid-Kuparuk region (K7), which is separated from the upper Kuparuk by another substantial dry zone, was strongly admixed with clusters 1–6. Cluster 4 (orange) dominated in the upper Sagavanirktok watershed (S1–4), whereas cluster 5 (maroon) dominated in the Oskrukuyik tributary of the Sagavanirktok (S5–6), above another substantial dry zone. The mid-Sagavanirktok region (S7) was dominated by cluster 6 (pink) with additional admixture, especially from the upper Sagavanirktok and Oksrukuyik clusters.

Discriminant analysis of principal components (DAPC) on microsatellite genotype data. Individuals are assigned to one of five genetic clusters, represented by both color and shape (black-open square, aqua-diamond, blue-circle, purple-triangle, and green-closed square). The shaded vertical bars are the eigenvalue histogram (proportion of conserved variance = 0.466)
DAPC analysis produced similar results for individual assignment probabilities to those found using STRUCTURE. However, DAPC assigned individuals from the Toolik region (K5 and K6), Lower Kuparuk region (K7) and Lower Sagavanirktok region (S7) to the same cluster. Both STRUCTURE and DAPC indicated genetic admixture for individuals from the Lower Sagavanirktok (K7) and Lower Kuparuk (K7) regions (Figs. 1 and 2).
Effective population size
Effective population size estimates for upstream headwater regions ranged from 168 to 551 individuals, including the Upper Sagavanirktok region (S1–S4) Ne = 458, Oksrukuyik region (S5, S6) Ne = 168, Upper Kuparuk region (K1 – K4) Ne = 551, Toolik region (K5 & K6) Ne = 213 and Upper Itkillik region (C1) Ne = 320. The effective population size for all downstream locations was not estimable (returning an arbitrarily large number, 99,999), which is a common outcome when population sizes are large and the signal of drift is small (Waples and Do 2010). Given that the same sample sizes from headwater populations returned sensible population estimates and the known behavior of these estimators for large populations, we cautiously suggest that downstream populations are larger in census size than upstream populations even if a precise estimate is not possible. Thus, estimates of effective population size (Ne) are likely larger for downstream locations (S7, K7, and C2) compared to upstream (headwater) locations (Fig. 3; Table S10), although more downstream sampling is needed to support this proposition.

Effective population size (NeESTIMATOR) and pairwise gene flow (BAYESASS3) (m ≥ 0.02 shown) among geographic regions (abbreviations as in Fig. 1; Table S1). Ne is indicated by median and 95% confidence intervals. Infinite or large values for Ne (99,999) indicate a large population size with a low signal of drift that prevents definitive estimates. Arrow direction and line-thicknesses represent direction and magnitude of gene flow (m), respectively (see Table S11 for standard deviations)
Recent pairwise gene flow
Analysis of pairwise rates of gene flow over the last two generations (m) provided bidirectional estimates among geographic regions (Fig. 3; Table S11). All model runs indicated convergence and varied little in model deviance. We present the model with the lowest deviance as suggested by Meirmans (2014). Pairwise gene flow estimates varied from m = 0–0.24 and was asymmetric within each major watershed (Fig. 3; Table S11). In the Sagavanirktok watershed, gene flow was downstream-biased with the highest rates of gene flow from headwater regions, Upper Sagavanirktok (S1–S4) and Oksrukuyik (S5 and S6), to the downstream region, Lower Sagavanirktok (S7). Similarly, within the Kuparuk watershed, downstream-biased gene flow occurred with high rates of gene flow from the headwater regions, Upper Kuparuk (K1–K4) and Toolik (K5 and K6), to the downstream region, Lower Kuparuk (K7). High gene flow within the Kuparuk watershed also occurred between the two sub-watershed regions (Upper Kuparuk (K1–K4) and Toolik (K5 and K6), with bias favoring gene flow from the Upper Kuparuk region to the Toolik region. The highest rate of gene flow occurred within the Colville/Itkillik watershed with upstream bias from the Ublutuoch region (C2) near the coast to the Upper Itkillik region (C1) near the headwaters.
Landscape genetics
We found that river distance and river dry zone distance significantly predicted 88% of the total genetic variation (Table 3). River distance accounted for 70% of total variance, and dry zone distance accounted for 18% of the total variance in genetic distance among locations. Elevation and coastal distance were not significant predictors of genetic distance among sampling locations. We obtained similar results from models that used alternative variables, including number of river dry zones rather than river dry zone distance and watershed association (from the same or a different watershed) rather than coastal distance, which underscored the importance of river distance and river dry zones for predicting genetic distance among sampling locations (Table S6A). Furthermore, Mantel and partial Mantel tests further indicated the significance of distance and river dry zones as predictor variables for explaining genetic differentiation among sampling locations (Table S6B; Fig. S7).
Discussion
This study suggests that geography and river drying can affect dispersal, gene flow, and genetic structure in a metapopulation of a key fish species in the Arctic. We found that North Slope Arctic grayling genetic structure was associated with both isolation-by-river distance and isolation-by-environment (river dry zones). Both factors were associated with restricted gene flow among populations, thereby facilitating drift and contributing to genetic differentiation among sampling locations. We also found asymmetric, downstream biased gene flow in watersheds containing river dry zones. Other genetic patterns, including lower allelic richness and heterozygosity in headwater regions compared to downstream regions add support for historical dispersal from glacial refugia or population isolation. In addition to being a genetically diverse ancestral population, the downstream flow of genetic variants that reach higher frequencies either through drift or selection in the headwater regions could also maintain genetic variation in these downstream regions.
Patterns of genetic diversity
We found significant isolation-by-river distance, which is consistent with results from other studies on Arctic grayling (Stamford and Taylor 2005; 2004; Reilly et al. 2014). Redenbach and Taylor (1999) suggested that North Slope Arctic grayling were probably confined to glacial refugia located North of the Brooks Range and extending along the Bering Coast. Our finding of higher genetic variability within larger downstream/coastal regions along with isolation by river distance supports this assertion. Similarly, Reilly et al. (2014) found that Arctic grayling heterozygosity and allelic richness were highest on the coastal plain and lower for headwater regions: a pattern that might indicate dispersal from ancient refugia in the north and subsequent colonization of upstream headwater locations to the south (Nei et al. 1975).
We also found low genetic differentiation and high gene flow between the Ublutuoch region on the coastal plain and our unimpeded headwater location within the Upper Itkillik tributary of the Colville River, suggesting that either the Arctic grayling’s ability to travel long distances might aid dispersal in the absence of gene flow barriers or that grayling might follow a stepping-stone model of dispersal (Kimura and Weiss 1964). Our understanding of grayling long-distance movement in unimpeded streams lends support to the former explanation (West et al. 1992). Adding to the growing recognition for other salmonid species (Kanno et al. 2011; Meeuwig et al. 2010; Poissant et al. 2005), we found fine-scale genetic differentiation in impeded watersheds containing dry river zones.
Dry zones as barriers to gene flow
Physical barriers often play a role in determining fine-scale genetic structure of freshwater fish species (Whiteley et al. 2006, 2010; Kanno et al. 2011; Junker et al. 2012; Junge et al. 2014; Kelson et al. 2015), but often the degree to which they disrupt gene flow depends largely on the dispersal ability of the species and the permeability of barriers (Bergerot et al. 2015). Arctic grayling are capable of long-distance movement both within and among watersheds (West et al. 1992), which could help explain genetic similarities between individuals from the Upper Itkillik and Ublutuoch regions, which were sampled 379 km apart. We have never observed dry zones along the Itkillik or Colville Rivers, suggesting that this system remains well-connected throughout the open-water season, thereby allowing fish to move freely from headwaters to coast without increased risk of becoming trapped or dying due to river desiccation. Interestingly, although dry zones are imperfect barriers and Arctic grayling has high dispersal capability, we also detected microgeographic genetic differentiation in our system at scales of less than 20 km.
Microgeographic genetic differentiation, like that observed in our system, occurs when population divergence exists within the dispersal range of the species and suggests that factors, such as barriers to gene flow or strong natural selection, might shape population structure (Richardson et al. 2014). River dry zones are unlikely to prevent movement, restrict access to mates, or present physical barriers to gene flow during the Arctic grayling mating season when gametes are exchanged because river discharge is often at its maximum during the spring freshet (Lammers et al. 2001). Admixed individuals found throughout our study system provide evidence that straying among regions and successful mating occurs. Thus, the mechanism by which dry zones influence genetic differentiation might not be as straightforward as physical isolation by a barrier, but rather might involve indirect restriction of gene flow, such as strong natural selection against maladapted migrants or low fitness (Gilk et al. 2004). Admixture can produce intermediate phenotypes maladapted to either parents’ native environment. For grayling, traits such as overwintering site selection might become maladapted for the admixed offspring of a parent that overwinters upstream in headwater lakes and a parent that overwinters downstream in coastal streams.
Drought-prone Arctic tundra streams might affect fish population genetic structure similarly to drought-prone desert aquatic systems, where stream distance and river drying best predicted genetic divergence among sites for desert fish (Fitzpatrick et al. 2014). In our study system, river drying often occurs when low precipitation and high evapotranspiration rates reduce stream flow (Betts and Kane 2015), conditions which most often occur during late summer when adult Arctic grayling must migrate to overwintering locations. During one such event, we witnessed Arctic grayling trapped by a dry zone in the Kuparuk River and found that trapped fish experienced overcrowding, decreased body condition, and increased predation—all factors that could promote strong selection. Further research is needed to determine if river dry zones associated with isolation-by-environment also influence Arctic grayling fitness and if fitness differences promote locally adapted phenotypes.
Asymmetric gene flow and effective population size
We detected asymmetric gene flow from small, semi-isolated headwater regions to larger, unobstructed coastal regions, which might be due to high discharge events favoring downstream movement or to higher environmental variability within headwater regions. Arctic grayling spawning migration occurs during springtime when river discharge is high, which might promote downstream dispersal from the headwaters toward the coast. Downstream displacement of juveniles and/or fertilized eggs might occur during high discharge events, increasing the potential for gene flow from headwaters to coast (Harvey 1999; Van Leeuwen et al. 2017). Another explanation involves increased emigration from the headwaters due to high environmental variability within those regions, such as fluctuating stream hydrology. Altermatt and Ebert (2010) suggested that environmental variability could promote dispersal from less to more stable conditions, as found for Daphnia magna in freshwater ephemeral pools along the Baltic Sea. They found that small populations in environmentally variable habitat patches produced proportionally more long-distance dispersers than large, long-lived populations. Such dynamics might arise when conditions affecting local survival, including habitat size, quality, and stability, negatively correlate with dispersal (Altermatt and Ebert 2010). In Arctic tundra streams, drought-prone headwater habitats might produce conditions that negatively correlate with Arctic grayling survival (i.e., warm temperatures, deoxygenation, desiccation), possibly promoting higher rates of gene flow from small headwater regions to larger coastal regions.
Although the rate of gene flow from headwater regions to coastal regions was higher than from the coast to the headwaters, the potential number of effective dispersers from the coast to the headwaters could be high due to potentially large size of the downstream populations. Ne estimates and confidence interval bounds tend to be more accurate for small populations and less accurate for large populations due to the disproportionately low influence of drift in large populations (Waples and Do 2010). This inequity helps explain the infinite results (i.e. Ne = 99,999) and large confidence intervals found for our downstream populations because Ne is difficult to measure when population size is large and drift is small. Interestingly, despite potentially high effective dispersal from the coast to the headwaters, finer scaled genetic differentiation within headwater regions suggests the possibility that other isolating mechanisms, such as pre-zygotic isolation (i.e. sexual selection) or post-zygotic isolation (i.e. low hybrid fitness), could be operating in this system.
Asymmetric gene flow, connectivity, and conservation
Our study indicates that semi-isolated upstream regions likely contribute to higher levels of genetic variation among headwater regions of the North Slope Arctic grayling metapopulation. Similar to observations by Morrissey and Kerekhove (2009) for other freshwater metapopulations, restricted upstream gene flow to small semi-isolated headwater populations can promote genetic drift along with the creation and maintenance of distinctive genotypes. The isolation of upstream habitats coincident with differences in environmental conditions among upstream locations (e.g., high predation levels in overwintering lakes (Buzby and Deegan 2004)) might facilitate local adaptation in these smaller populations (Funk et al. 2012). Downstream-biased gene flow might transport higher frequencies of these genetic variants downstream, although we cannot distinguish this effect from the existing diversity found in the larger downstream ancestral population. Downstream gene flow combined with the higher genetic diversity in the large, ancestral population might create genetic reservoirs of adapted alleles which could, in turn, promote species persistence through redistribution and recolonization by downstream individuals into upstream habitat patches following disturbances (Rieman and Dunham 2000).
Although climate change might increase local extirpation risks, high overall genetic diversity across the metapopulation might enhance resilience, which could promote population persistence as long as some level of connectivity persists. However, maintaining aquatic connectivity might become challenging if Arctic summers continue to warm and the frequency and duration of river drying increase (Kane et al. 2004; Hinzman et al. 2005; Betts and Kane 2015). Increased river drying could decrease dispersal and further isolate populations. Without re-colonization, stochastic events might eradicate some of these small headwater populations. Ideally, the management of Arctic grayling populations in a warming climate should include an understanding of the relative strengths of gene flow, drift, and population demographic rates, which for Arctic grayling all hinge upon aquatic connectivity.
Overall, we need to understand better how altered connectivity influences genetic structure and either promotes resilience or extirpation (Campbell Grant et al. 2007; Fagan 2002; Labonne et al. 2008), especially in systems with asymmetric gene flow. In aquatic systems, upstream populations are likely to be isolated and act as cradles of higher frequencies of potentially adaptive alleles, whereas downstream populations tend to be larger and more genetically diverse. Downstream populations might act as reservoirs of genetic diversity, facilitating their resistance to future change. However, upstream populations are likely to become increasingly isolated, threatening both the population's persistence and its store of adaptive alleles (Rieman and Dunham 2000). Preserving the full breadth of genetic diversity in a species thus likely requires maintaining some level of upstream connectivity for downstream genetic reservoirs to function as effective genetic and conservation safety nets.
Data availability
All data will be deposited in a public data repository on publication.
References
ACIA (2005) Arctic Climate Impact Assessment. ACIA Overview report. Cambridge University Press
Altermatt F, Ebert D (2010) Populations in small, ephemeral habitat patches may drive dynamics in a Daphnia magna metapopulation. Ecology 91:2975–2982
Beauchamp DA (1990) Movements, habitat use, and spawning strategies of arctic grayling in a subalpine lake tributary. Northwest Sci 64:195–207
Bergerot B, Hugueny B, Belliard J (2015) Relating life-history traits, environmental constraints and local extinctions in river fish. Freshw Biol 60:1279–1291. https://doi.org/10.1111/fwb.12561
Betts ED, Kane DL (2015) Linking north slope of Alaska climate, hydrology, and fish migration. Hydrol Res. https://doi.org/10.2166/nh.2014.031
Blair SD, Matheson D, He Y, Goss GG (2016) Reduced salinity tolerance in the Arctic grayling (Thymallus arcticus) is associated with rapid development of a gill interlamellar cell mass: implications of high-saline spills on native freshwater salmonids. Conserv Physiol 4(1):cow010
Buzby KM, Deegan LA (2004) Long-term survival of adult Arctic grayling (Thymallus arcticus) in the Kuparuk river, Alaska. Can J Fish Aquat Sci 61:1954–1964
Campbell Grant EH, Lowe WH, Fagan WF (2007) Living in the branches: population dynamics and ecological processes in dendritic networks. Ecol Lett. https://doi.org/10.1111/j.1461-0248.2006.01007.x
Diggs MD, Ardren WR (2008) Characterization of 12 highly variable tetranucleotide microsatellite loci for Arctic grayling (Thymallus arcticus) and cross amplification in other Thymallus species. Mol Ecol Resour. https://doi.org/10.1111/j.1755-0998.2007.02081.x
Do C, Waples RS, Peel D, Macbeth GM, Tillett BJ, Ovenden JR (2014) NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour. https://doi.org/10.1111/1755-0998.12157
Earl DA, VonHoldt BM (2011) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361. https://doi.org/10.1007/s12686-011-9548-7
England PR, Cornuet J-M, Berthier P, Tallmon DA, Luikart G (2006) Estimating effective population size from linkage disequilibrium: severe bias in small samples. Conserv Genet 7(2):303–308. https://doi.org/10.1007/s10592-005-9103-8
ESRI (2013) ArcGIS desktop: release 10.2. Environmental Systems Research Institute, Redlands, CA
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Fagan W (2002) Connectivity, fragmentation, and extinction risk in dendritic metapopulations. Ecology 83:3243–3249
Fagan WF, Unmack PJ, Burgess C, Minckley WL (2007) Rarity, fragmentation, and extinction risk in desert fishes. Ecology. https://doi.org/10.1890/0012-9658(2002)083%5B3243:CFAERI%5D2.0.CO%3B2
Faubet P, Waples R, Gaggiotti O (2007) Evaluating the performance of a multilocus Bayesian method for the estimation of migration rates. Mol Ecol 16:1149–1166. https://doi.org/10.1111/j.1365-294X.2007.03218.x
Fausch KD, Torgersen CE, Baxter CV, Li HW (2002) Landscapes to riverscapes: bridging the gap between research and conservation of stream fishes. Bioscience 52:483–498
Fitzpatrick SW, Crockett H, Funk WC (2014) Water availability strongly impacts population genetic patterns of an imperiled Great Plains endemic fish. Conserv Genet. https://doi.org/10.1007/s10592-014-0577-0
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol. https://doi.org/10.1016/j.tree.2012.05.012
Galpern P, Peres-Neto P, Polfus J, Manseau M (2014) MEMGENE: spatial pattern detection in genetic distance data. Methods Ecol Evol. https://doi.org/10.1111/2041-210X.12240
Gilk SE, Wang IA, Hoover CL, Smoker WW, Taylor SG, Gray AK, Gharrett AJ (2004) Outbreeding depression in hybrids between spatially separated pink salmon, onchorhynchus gorbuscha, populations: marine survival, homing ability, and variability in family size. Environ Biol Fishes 69:287–297. https://doi.org/10.1023/B:EBFI.0000022888.28218.c1
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics. https://doi.org/10.2307/2532296
Hamilton TD (2003) Glacial geology of the toolik lake and upper kuparuk river regions (No. 26). In D Walker (ed) Biological papers of the University of Alaska, Fairbanks, Alaska
Harvey BC (1999) Susceptibility of young-of-the-year fishes to downstream displacement by flooding. Trans Am Fish Soc. https://doi.org/10.1577/1548-8659(1987)116%3c851:SOYFTD%3e2.0.CO;2
Hinzman LD, Bettez ND, Bolton WR et al (2005) Evidence and implications of recent climate change in Northern Alaska and other Arctic regions. Climatic Change. https://doi.org/10.1007/s10584-005-5352-2
Hoffmann AA, Sgrò CM (2011) Climate change and evolutionary adaptation. Nature. https://doi.org/10.1038/nature09670
Hopken MW, Douglas MR, Douglas ME (2013) Stream hierarchy defines riverscape genetics of a North American desert fish. Mol Ecol. https://doi.org/10.1111/mec.12156
IPCC (2013) Climate change 2013: the physical science basis. IPCC, Geneva, Switzerland
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. https://doi.org/10.1093/bioinformatics/btm233
Jenkins DG, Carey M, Czerniewska J, Fletcher J, Hether T, Jones A, Knight S, Knox J, Long T, Mannino M, McGuire M, Riffle A, Segelsky S, Shappell L, Sterner A, Strickler T, Rursi R (2010) A meta-analysis of isolation by distance: relic or reference standard for landscape genetics? Ecography 33:315–320. https://doi.org/10.1111/j.1600-0587.2010.06285.x
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics. http://www.biomedcentral.com/1471-2156/11/94.
Junge C, Museth J, Hindar K, Kraabøl M, Vøllestad LA (2014) Assessing the consequences of habitat fragmentation for two migratory salmonid fishes. Aquat Conserv. https://doi.org/10.1002/aqc.2391
Junker J, Peter A, Wagner CE, Mwaiko S, Germann B, Seehausen O, Keller I (2012) River fragmentation increases localized population genetic structure and enhances asymmetry of dispersal in bullhead (Cottus gobio). Conserv Genet. https://doi.org/10.1007/s10592-011-0306-x
Kalinowski ST (2005) Hp-Rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Kamvar ZN, Tabima JF, Grünwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kane DL, Gieck RE, Kitover DC, Hinzman LD, McNamara JP, Yang D (2004) Hydrologic cycles on the North Slope of Alaska. In: Kane DL, Yang D (eds) Northern research basins water balance. IAHS Press, Wallingford, UK, pp 224–236
Kanno Y, Vokoun JC, Letcher BH (2011) Fine-scale population structure and riverscape genetics of brook trout (Salvelinus fontinalis) distributed continuously along headwater channel networks. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2011.05210.x
Kelson SJ, Kapuscinski AR, Timmins D, Ardren WR (2015) Fine-scale genetic structure of brook trout in a dendritic stream network. Conserv Genet. https://doi.org/10.1007/s10592-014-0637-5
Kimura M, Weiss GH (1964) The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics. https://doi.org/10.1093/oxfordjournals.molbev.a025590
Labonne J, Ravigne V, Parisi B, Gaucherel C (2008) Linking dendritic network structures to population demogenetics: the downside of connectivity. Oikos. https://doi.org/10.1111/j.2008.0030-1299.16976.x
Lammers RB, Shiklomanov AI, Vorosmarty CJ, Fekete BM, Peterson BJ (2001) Assessment of contemporary Arctic river runoff based on observational discharge records. J Geophys Res 106:3321–2224. https://doi.org/10.1029/2000JD900444
Li Y, Liu J (2018) Structure selector: a web-based software to select and visualize the optimal number of clusters using multiple methods. Mol Ecol Res 18(1):176–177. https://doi.org/10.1111/1755-0998.12719
Lowe WH, Likens GE, McPeek MA, Buso DC (2006) Linking direct and indirect data on dispersal: Isolation by slope in a headwater stream salamander. Ecology 87(2):334–339. https://doi.org/10.1890/05-0232
Martin PD, Jenkins JL, Adams FJ, et al (2009) Wildlife response to environmental arctic change: predicting future habitats of Arctic Alaska. Report of the wildlife response to environmental arctic change (WildREACH): predicting future habitats of Arctic Alaska Workshop, Fairbanks, Alaska
Manel S, Holderegger R (2013) Ten years of landscape genetics. Trends Ecol Evol. https://doi.org/10.1016/j.tree.2013.05.012
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
Maria ALP, Irene K, Anja Marie W, Christopher TR (2012) How river structure and biological traits influence gene flow: a population genetic study of two stream invertebrates with differing dispersal abilities. Freshw Biol. https://doi.org/10.1111/j.1365-2427.2012.02758.x
Meeuwig MH, Guy CS, Kalinowski ST, Fredenberg WA (2010) Landscape influences on genetic differentiation among bull trout populations in a stream-lake network. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2010.04655.x
Meffe GK, Vrijenhoek RC (1988) Conservation genetics in the management of desert fishes. Conserv Biol 2(2):157–169. https://doi.org/10.1111/j.1523-1739.1988.tb00167.x
Meirmans PG (2014) Nonconvergence in Bayesian estmation of migration rates. Mol Ecol Res 14(4):726–733. https://doi.org/10.1111/1755-0998.12216
Meirmans PG, Van Tienderen PH (2004) GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Morrissey MB, de Kerckhove DT (2009) The maintenance of genetic variation due to asymmetric gene flow in dendritic metapopulations. Am Nat. https://doi.org/10.1086/599011
Mossop KD, Adams M, Unmack PJ, Smith Date KL, Wong BBM, Chapple DG (2015) Dispersal in the desert: ephemeral water drives connectivity and phylogeography of an arid-adapted fish. J Biogeogr. https://doi.org/10.1111/jbi.12596
Nei M, Li W (1973) Linkage disequilibrium in subdivided populations. Genetics 75(1):213–219
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution. http://www.jstor.org/stable/2407137.
Nomura T (2008) Estimation of effective number of breeders from molecular coancestry of single cohort sample. Evol Appl. https://doi.org/10.1111/j.1752-4571.2008.00015.x
Northcote TG (1995) Comparative biology and management of Arctic and European grayling (Salmonidae, Thymallus). Rev Fish Biol Fish 5(2):141–194. https://doi.org/10.1007/bf00179755
Oksanen J, Blanchet FG, Kindt R et al (2013) Vegan: community ecology package. SAGE Publications Inc., California. https://doi.org/10.4135/9781412971874.n145
Opdam P, Wascher D (2004) Climate change meets habitat fragmentation: linking landscape and biogeographical scale levels in research and conservation. Biol Cons. https://doi.org/10.1016/j.biocon.2003.12.008
Ozerov MY, Veselov AE, Lumme J, Primmer CR, Moran P (2012) “Riverscape” genetics: river characteristics influence the genetic structure and diversity of anadromous and freshwater Atlantic salmon (Salmo salar) populations in northwest Russia. Can J Fish Aquat Sci. https://doi.org/10.1139/f2012-114
Parkinson D, Philippart JC, Baras E (1999) A preliminary investigation of spawning migrations of grayling in a small stream as determined by radio-tracking. J Fish Biol 55:172–182
Parmesan C (2006) Ecological and evolutionary responses to recent climate change. Annu Rev Ecol Evol Syst 37:637–669
Perkin JS, Gido KB, Costigan KH, Daniels MD, Johnson ER (2015) Fragmentation and drying ratchet down Great Plains stream fish diversity. Aquat Conserv Marine Freshw Ecosyst. https://doi.org/10.1002/aqc.2501
Peterson DP, Ardren WR (2009) Ancestry, population structure, and conservation genetics of Arctic grayling (Thymallus arcticus) in the upper Missouri river, USA. Can J Fish Aquat Sci. https://doi.org/10.1139/F09-113
Peterson EE, Ver Hoef JM (2014) STARS: an ArcGIS toolset used to calculate the statistical models to stream network data. J Stat Softw 56:1–17
Poissant J, Knight TW, Ferguson MM (2005) Nonequilibrium conditions following landscape rearrangement: the relative contribution of past and current hydrological landscapes on the genetic structure of a stream-dwelling fish. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2005.02500.x
Primmer CR, Veselov AJ, Zubchenko A, Poututkin A, Bakhmet I, Koskinen MT (2006) Isolation by distance within a river system: genetic population structuring of Atlantic salmon, Salmo salar, in tributaries of the Varzuga river in northwest Russia. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2005.02844.x
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics. http://www.genetics.org/cgi/content/abstract/155/2/945.
Puechmaille SJ (2016) The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627
Rambaut A, Drummond AJ, Suchard M (2003) Tracer v1.6: MCMC trace analysis package. Institute of Evolutionary Biology, Department of Computer Science. University of Edinburgh, Edinburgh
Rannala B (2007) BayesAss edition 3.0 user’s manual. Department of Evolution and Ecology, University of California, Davis, CA
Redenbach Z, Taylor EB (1999) Zoogeographical implications of variation in mitochondrial DNA of Artic grayling (Thymallus arcticus). Mol Ecol 8:23–35
Reilly JR, Paszkowski CA, Coltman DW (2014) Population genetics of Arctic grayling distributed across large, unobstructed river systems. Trans Am Fish Soc. https://doi.org/10.1080/00028487.2014.886620
Reist JD, Wrona FJ, Prowse TD, Power M, Dempson JB, King JR, Beamish RJ (2006) An overview of effects of climate change on selected arctic freshwater and anadromous fishes. Ambio 35:381–387
Richardson JL (2012) Divergent landscape effects on population connectivity in two co-occurring amphibian species. Mol Ecol 21:4437–4451
Richardson JL, Urban MC, Bolnick DI, Skelly DK (2014) Microgeographic adaptation and the spatial scale of evolution. Trends Ecol Evol. https://doi.org/10.1016/j.tree.2014.01.002
Rieman BE, Dunham JB (2000) Metapopulations and salmonids: a synthesis of life history patterns and empirical observations. Ecol Freshw Fish. https://doi.org/10.1034/j.1600-0633.2000.90106.x
Roberts JH, Angermeier PL, Hallerman EM (2013) Distance, dams and drift: what structures populations of an endangered, benthic stream fish? Freshw Biol. https://doi.org/10.1111/fwb.12190
Rosenberg NA (2003) Distruct: a program for the graphical display of population structure. Mol Ecol Notes. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENPOP software for Windows and Linux. Mol Ecol Resour. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Scheffers BR, De Meester L, Bridge TCL, Hoffmann AA, Pandolfi JM, Corlett RT, Butchart SHM, Pearce-Kelly P, Kovacs KM, Dudgeon D, Pacifici M, Rondinini C, Foden WB, Martin TG, Mora C, Bickford D, Watson JEM (2016) The broad footprint of climate change from genes to biomes to people. Science 354:aaf7671
Schloss CA, Nuñez TA, Lawler JJ (2012) Dispersal will limit ability of mammals to track climate change in the Western Hemisphere. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1116791109
SDMI (2013) Alaska Mapped Statewide Digital Mapping (SDMI). http://www.alaskamapped.org/. Accessed 1 Jan 2014
Stamford MD, Taylor EB (2004) Phylogeographical lineages of Arctic grayling (Thymallus arcticus) in North America: divergence, origins and affinities with Eurasian Thymallus. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2004.02174.x
Stamford MD, Taylor EB (2005) Population subdivision and genetic signatures of demographic changes in Arctic grayling (Thymallus arcticus ) from an impounded watershed. Can J Fish Aquat Sci. https://doi.org/10.1139/f05-156
Steed AC (2007) Population viability of Arctic grayling (Thymallus arcticus) in the Gibbon River. Montana State University, Yellowstone National Park
Storfer A, Murphy MA, Spear SF, Holderegger R, Waits LP (2010) Landscape genetics: where are we now? Mol Ecol. https://doi.org/10.1111/j.1365-294X.2010.04691.x
Urban MC, Zarnetske PL, Skelly DK (2013) Moving forward: dispersal and species interactions determine biotic responses to climate change. Ann N Y Acad Sci. https://doi.org/10.1111/nyas.12184
Urban MC, Bocedi G, Hendry AP et al (2016) Improving the forecast for biodiversity under climate change. Science. https://doi.org/10.1126/science.aad8466
USGS (2014) US geological survey. http://nhd.usgs.gov/. Accessed 1 Jan 2014
Vähä JP, Erkinaro J, Niemelä E, Primmer CR (2007) Life-history and habitat features influence the within-river genetic structure of Atlantic salmon. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2007.03329.x
Van Leeuwen CHA, Dokk T, Haugen TO, Kiffney PM, Museth J (2017) Small larvae in large rivers: observations on downstream mofement of European grayling Thymallus thymallus during early life stages. J Fish Biol 90(6):2412–2424. https://doi.org/10.1111/jfb.13326
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Wahlund S (1928) Zusammensetzung von populationen und korrelationserscheinungen vom standpunkt der vererbungslehre aus betrachtet. Hereditas. https://doi.org/10.1111/j.1601-5223.1928.tb02483.x
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl. https://doi.org/10.1111/j.1752-4571.2009.00104.x
Waples RS, Antao T, Luikart G (2014) Effects of overlapping generations on linkage disequilibrium estimates of effective population size. Genetics. https://doi.org/10.1534/genetics.114.164822
West RL, Smith MW, Barber WE, Reynolds JB, Hop H (1992) Autumn migration and overwintering of Arctic grayling in coastal streams of the Arctic national wildlife refuge. Transactions of the American Fisheries Society, Alaska. https://doi.org/10.1577/1548-8659(1992)121%3c0709:AMAOOA%3e2.3.CO;2
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics. http://www.genetics.org/cgi/content/abstract/163/3/1177.
Whiteley AR, Spruell P, Rieman BE, Allendorf FW (2006) Fine-scale genetic structure of bull trout at the southern limit of their distribution. Trans Am Fish Soc. https://doi.org/10.1577/T05-166.1
Whiteley AR, Hastings K, Wenburg JK, Frissell CA, Martin JC, Allendorf FW (2010) Genetic variation and effective population size in isolated populations of coastal cutthroat trout. Conserv Genet. https://doi.org/10.1007/s10592-010-0083-y
Whitlock MC (2011) G’ST and D do not replace FST. Mol Ecol. https://doi.org/10.1111/j.1365-294X.2010.04996.x
Acknowledgements
Research was funded by National Science Foundation Award PLR-1417754. MCU was also supported by the Center of Biological Risk and Arden Chair in Ecology and Evolutionary Biology, and HEG was supported by the US Environmental Protection Agency (EPA) STAR fellowship (EPA STAR F91745901).
Funding
Provided by NSF award PLR-1417754 and EPA STAR FP91745901.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they have no conflicts of interest.
Ethical approval
Research was performed under IACUC protocol 17–18.
Consent for publication
All authors have given consent for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Golden, H.E., Holsinger, K.E., Deegan, L.A. et al. River drying influences genetic variation and population structure in an Arctic freshwater fish. Conserv Genet 22, 369–382 (2021). https://doi.org/10.1007/s10592-021-01339-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-021-01339-0