
Abstract
Hispaniola is the second largest island in the Caribbean and harbors an extensive amount of biodiversity. The geologic history and resulting complex topography of the island has led to significant differentiation across various taxonomic groups. Hispaniola is the only Caribbean Island with two species of Rock Iguanas, genus Cyclura. Rhinoceros Rock Iguanas (C. cornuta) are wide-ranging across Hispaniola, occurring in isolated pockets, primarily in low elevation xeric areas. To better understand the population structure of this species, we used a combination of mtDNA and nuclear markers to elucidate the genetic variation of wild populations across 13 sampling regions in the Dominican Republic (DR), as well as neighboring Mona Island, home to a Cyclura population of uncertain taxonomic status. Further, we evaluate the origin of iguanas in captive facilities throughout the DR. Our data reveal a high degree of genetic diversity across wild populations within the DR and shed light on the taxonomic status of the Mona island population. Further, novel genetic diversity is found in captive facilities, most likely resulting from interbreeding between individuals from genetically distinct populations within the captive facilities. Our results suggest that the captive facilities may pose a threat to wild populations and increased regulation of these facilities is needed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The continuous and detrimental influx of human activities on natural habitats demands conservation actions that integrate population genetics and evolutionary biology into management decisions (Ralls et al. 2017). To address population decline and fragmentation with effective management strategies, accurate information regarding population genetic structure, gene flow, geographic barriers, and time in isolation is required. However, this information is often lacking and predicting patterns of local adaptation, inbreeding depression, and/or outbreeding depression is increasingly difficult in long-lived organisms (Taylor et al. 2017), making the selection of appropriate conservation strategies daunting. Assessing intraspecific variation remaining in the wild and characterizing population differences should be among the primary goals for conservation geneticists interested in contributing to management decisions (Waples 1998; Avise 2005; Pasachnik et al. 2011).
Populations ranging over large areas are likely to present significant genetic and morphological variation. This differentiation frequently correlates with the presence of conspicuous barriers to gene flow and migration, such as mountains, waterways, or simply patches of unsuitable habitat. These patterns of differentiation are not only expected on continental mainland but also within large islands, especially when geographic barriers were historically present.
Hispaniola is the second largest island in the Greater Antilles and ranks second in Caribbean biodiversity (Ministerio de Medio Ambiente y Recursos Naturales 2014). The island was formed when two paleo-island blocks collided in the mid-Miocene, ca. 10 mya. Major orogenic events ceased during this time creating the stable island topography known today (Mann et al. 1991; Iturralde-Vinent and MacPhee 1999). Hispaniola has five major mountain ranges covering most of the island and creating an extensive system of natural barriers (Fig. 1, Nairn and Stehli 1975). Further, low elevation areas, most notably the Enriquillo Basin, at the north and south paleo-island boundary, continued to submerge multiple times until the late Pleistocene (Maurasse 1982; Graham 2003), serving as barriers to dispersal for terrestrial fauna (Gifford et al. 2004; Gifford and Larson 2008; Glor and Warren 2011; Brace et al. 2012). These geographic barriers have throughout time contributed to the distribution of genetic variation in reptiles, such as Ameiva chrysolaema (now Pholidoscelis chrysolaema) (Gifford and Larson 2008; Gifford et al. 2004), Chilabothrus striatus (Reynolds et al. in prep), and Cyclura ricordii (Carreras De León et al. 2019), as well as other vertebrates (Townsend et al. 2007; Sly et al. 2011; Brace et al. 2012).

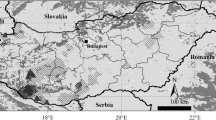
Notable geographic features (mountains and valleys) in the Dominican Republic. Topology is represented with lighter shades indicating higher elevation. Blue indicates water areas. The dashed brown line represents the historical collision site between the northern and southern paleo-islands, forming Hispaniola. The location of 18 sampled iguanarios within the Dominican Republic is represented with yellow dots
Cyclura cornuta is one of two species of Rock Iguanas endemic to Hispaniola. This species has a widespread distribution across the island but is primarily found in low elevation tropical dry forest habitats, and never on high elevation mountains. Recent surveys indicate that this species may be much less stable than previously thought (Pasachnik and Carreras De León 2014), and an updated IUCN Red List Assessment supports its current Endangered status (Pasachnik and Carreras De León 2019). The primary threats to this species are habitat degradation, invasive species, and harvesting for human use (Rupp et al. 2009; Rupp and León 2009). More recently Pasachnik and Carreras-De León (2014) identified iguanarios, or captive facilities, as a novel potential threat to C. cornuta within the Dominican Republic (DR).
These captive facilities range from small (1–5 individuals) to large scale (> 100 individuals) and are distributed across the DR with greatest prevalence in the eastern portion of the country (Pasachnik and Carreras De León 2014). The first iguanario, Manati Park, was created in 1997 to help alleviate an overabundance of confiscated iguanas at the National Zoo in Santo Domingo, DR (ZooDom). In the 1990’s it was also believed that iguanarios could serve as education centers and breeding source populations, in time supplementing wild populations (Powell et al. 2002). Augmenting declining populations with individuals bred in captive or semi-captive conditions is a conservation strategy that has been widely used (Grativol et al. 2001; Searcy et al. 2009; Frankham et al. 2010; Grant and Hudson 2015). However, in order for these strategies to be successful, detailed information concerning genetics and distribution must be used to create a long-term management plan (Frankham et al. 2010). In the case of iguanarios in the DR, little to no record keeping has occurred, animals are not given a unique identifier, the movement of individuals between facilities is not recorded, reproduction is not managed, and there is no overarching plan by which the facilities function together. Breeding has been extremely successful in the larger facilities and most now have an overabundance of iguanas. Further, growing tourist interest in these sites has triggered the creation of additional iguanarios in the southeastern portion of the country, where tourism is most prominent. The condition in many of these facilities is very poor, due to overcrowding and a lack of proper husbandry knowledge. As a rapid solution, iguanas are often released haphazardly into the surrounding area or given away to create new facilities. In addition, animals sometimes escape, due to poor security measures (Pasachnik and Carreras De León 2014).
There is very little information available concerning the genetic variability of C. cornuta in the wild (Malone et al. 2000), and virtually no data on the individuals currently kept in iguanarios across the DR (Pasachnik and Carreras De León 2014). The potential of using iguanarios as an effective conservation strategy is not viable under the current conditions. Instead, the situation may now pose a threat to the genetic integrity and health of the wild populations through the potential loss of local adaptation and disease introduction (Pasachnik and Carreras De León 2014).
In this paper, we aim to elucidate the overall population genetic structure of wild C. cornuta iguanas across the DR and test the hypothesis that vicariant events may have contributed to local differentiation of the species in the wild. Specifically, we predict that high elevation mountain ranges as well as low lying areas with frequent sea inundation have contributed to population isolation and subsequent differentiation. We then use the information from the wild populations to reconstruct the possible origin of captive individuals and guide proper management strategies for iguanarios. Further, while elucidating the population genetic structure of C. cornuta in the Dominican Republic, we aim to address the long-standing issue of the taxonomic status of Rock Iguanas on Mona Island. Mona is a small island in the Mona Channel, residing approximately 40 km to the east of Hispaniola and 40 km to the west of Puerto Rico. The population of Rock Iguanas on Mona has a close phenotypic resemblance to C. cornuta on Hispaniola and has been considered both a subspecies of C. cornuta (e.g., Schwartz and Carey 1977) as well as a separate species, Cyclura stejnegeri (Barbour and Noble 1916; see ITWG 2016 for an overview).
Material and methods
Field collection
DNA samples were collected from wild and captive individuals across the DR from 2010 to 2016 (Figs. 1, 2, 3). Wild iguana samples were collected across 13 regions of the DR (Figs. 2, 3; Table 1). The exact locations are not presented herein due to the threatened status of this species. The authors, upon legitimate request, may provide additional information. Samples were collected from Mona Island by Néstor Pérez Buitrago and loaned to San Diego Zoo Global Institute for Conservation Research. Captive samples were collected from 18 iguanarios (Fig. 1). In larger facilities samples were collected from a random subset of individuals. At the time of captive sample collection, informal interviews were conducted with the facility managers to obtain information regarding the history of the facility, the source of their iguanas, and the movement of iguanas from their facility (see Pasachnik and Carreras De León 2014 for a summary of this information). Samples could not be obtained from Haiti due to permitting obstacles. We collected samples from a total of 144 wild and 114 captive individuals.

Mitochondrial DNA haplotype distribution in wild individuals from the Dominican Republic and Mona Island. The size of the circle represents the number of samples in a given location as shown in the figure key. Numbers within circles represent sampling area number. Topology is represented with lighter shades indicating higher elevation. Pale blue areas indicate water

Distribution of nuclear DNA variability according to the DAPC analysis in wild individuals from the Dominican Republic and Mona Island. The size of the circle represents the number of samples in a given location as shown in the figure key. Numbers within circles represent sampling area number. Topology is represented with lighter shades indicating higher elevation. Pale blue areas indicate water
Upon capture of all wild and most captive individuals, snout-vent length, tail length, mass, and sex were recorded. To avoid re-sampling a passive integrated transponder (PIT) tag was implanted subcutaneously for permanent identification in all wild and some captive individuals. In addition, some individuals received bead tags (Rodda et al. 1988) for rapid identification. A 0.5 ml sample of blood was collected, from the caudal vein, from all live individuals. Blood was stored in EDTA buffer (Longmire et al. 1997). Tissue, collected from deceased individuals, was stored in 100% ETOH.
DNA extraction and genotyping
Genomic DNA was extracted using the Qiagen DNeasy DNA extraction kit (QIAGEN®). To assess mitochondrial DNA variation the polymerase chain reaction (PCR) was used to amplify a 674 bp portion of NADH dehydrogenase subunit 4 (see Pasachnik et al. 2009 for detailed procedures and primers). PCR products were verified by gel electrophoresis and successful amplicons were purified using exonuclease I/shrimp alkaline phosphate (ExoSAP). Sequencing reactions were preformed using original PCR primers by Eton Bioscience Inc. (San Diego, CA, USA).
Nuclear DNA variation was assessed using 18 microsatellites developed and used in previous studies involving Cyclura iguanas (see Table 2 for details). Double stranded DNA was quantified using Promega QuantiFluor® dsDNA System (Promega®) and the Thermo Fluoroskan Ascent Fluorometer (Thermo Scientific™). The extracted DNA was then normalized to 5 ng/µL. All forward primers were labeled with a fluorescent tag (6FAM, NED, VIC, or PET) attached to the 5′ end. Multiplexed PCR was run using five tagged primer sets per panel, the Qiagen Multiplex PCR mix (1X final concentration; QIAGEN®), and 20 ng of DNA. PCR cycling parameters included a 15-min hot start at 95 °C, followed by 40 cycles of 95 °C for 30 s, 60 °C annealing temperature for 45 s, and extension at 72 °C for 45 s. PCR products were diluted to an appropriate concentration determined by dilution tests. One microliter of diluted PCR product was added to 10 μl of HiDi Formamide with the 500 MW LIZ size standard (Applied Biosystems®) and 7µL of molecular grade water. Samples were analyzed on an ABI Prism 3730 DNA Analyzer (Applied Biosystems®) and data were called using GeneMarker® (Softgenetics®). Sample normalization, PCR, and preliminary analysis were performed at the University of Nevada, Nevada Genomics Center (https://www.ag.unr.edu/genomics/).
Data analysis
Wild genetic diversity and distribution
Sequences were trimmed and aligned using Sequencher® 4.6 (Gene Code Corporation, Ann Arbor, MI, USA). Ambiguous base calls were verified manually by examining the forward and reverse electropherogram reads. DnaSP v5 (Librado and Rozas 2009) was used to investigate the total number of haplotypes in our samples. Individual haplotypes were then grouped and visualized based on their geographic origin. Data were analyzed within the R statistical environment (R Development Core Team 2011) to test different hypothesis concerning geographic and genetic differentiation via a hierarchical Analysis of Molecular Variance (AMOVA). The R package poppr v2.5.0 (Kamvar et al. 2013) was used to run an AMOVA following Excoffier et al. (1992). The significance of our results was tested using a randomization test with 10,000 replicates in the R package ade4 v1.7.10 (Dray and Dufour 2007). Finally, identified haplotypes were used to reconstruct a phylogenetic network using the median joining algorithm implemented in the program PopART (Bandelt et al. 1999; Leigh and Bryant 2015).
Homozygous genotypes can sometimes be overrepresented when the frequency of null alleles in the dataset is too high. To estimate the frequency of null alleles in our dataset we used FreeNA (Dempster et al. 1977; Chapuis and Estoup 2007), and we set the null allele cut-off frequency to 0.2 as suggested by Chapuis and Estoup (2007). For individuals sampled at each independent location we further estimated a fixation index, observed heterozygosity, and expected heterozygosity (FIS, Ho, and He respectively) using Arlequin v3.5 (Excoffier et al. 1992, 2010). Additionally, we used the R package hierfstat (Goudet 2005) to calculate allelic richness, an estimate of the number of alleles rarefied by the number of individuals analyzed. To investigate patterns of differentiation across sampled regions we used microsatellite alleles to build a pairwise comparison of differences expressed using the FST index as calculated in Arlequin v3.5 (Excoffier et al. 2010). We then tested for significance across these comparisons with 1,000 permutations. Given the small sample sizes in some locations we grouped sites 3 and 4, 5 and 6, and 11–13, for these analyses based on geography and the STRUCTURE results (see below).
Microsatellite data are generally considered neutral to natural selection, but can hint to adaptive divergence when in tight linkage with regions of the genome directly under selection (Ellegren 2004; Edelist et al. 2006). To investigate the possibility of adaptive differences in wild populations of C. cornuta we used the microsatellite data in BAYESCAN v2.1 (Foll and Gaggiotti 2008). The software analyzes differences in allele frequencies across subpopulations following a model of isolation after splitting from an ancestral population. It then uses a fully Bayesian approach to estimate the posterior probability that differences in overall FST values between subpopulations could be caused by population specific effects (randomly evolving subpopulations) or by the interaction of locus and population specific effects (Foll and Gaggiotti 2008).
Bayesian based clustering algorithms are widely used in the analysis of population structure. To investigate the population genetic structure of wild Cyclura cornuta iguanas across the DR and Mona Island we used the software STRUCTURE v.2.3.4 (Pritchard et al. 2000; Falush et al. 2007). This software groups individuals according to a model of genetic distribution. We were interested in understanding the number of assigned genetic clusters (K) most explicative of our samples. The STRUCTURE software is known to present some biases when sample sizes are uneven across sampling locations (Puechmaille 2016). To address this issue, we limited the sample size from each sampled site to a maximum of 15 randomly selected individuals. Specifically, in regions 4 and 14, we collected 24 and 30 individuals respectively (Table 1). Therefore, for these sites, we randomly selected 15 individuals and ran the STRUCTURE analysis with a total of 120 wild samples. The simulation was run using a flat prior for the admixture parameter and using correlated allele frequencies. We set the program to test values of K ranging from 1 to 13. For each value of K the program performed 20 replicates of the analysis (Gilbert et al. 2012). In each independent analysis, Structure ran 106 MCMC while discarding the first 105 as burn-in. The final results were used to calculate the second order of differences in the likelihood function of K [ΔK] as implemented in the online software Structure-Harvester (Evanno et al. 2005; Earl and VonHoldt 2012). We used a bar-plot to represent relative probability of membership to assigned clusters.
We also investigated the population genetic structure with a Discriminant Analysis of Principal Components (DAPC) (Jombart et al. 2010). This multivariate analysis complements Bayesian clustering algorithms, such as STRUCTURE, because it doesn’t rely on any a priori model or assumption. The DAPC methodology partitions the overall genetic variance between- and within-groups, and looks for those variables maximizing the former while reducing the latter (Jombart et al. 2010).
To perform a DAPC we selected the same individuals used in the STRUCTURE analysis and used the R package adegenet, and its dependencies (Jombart 2008). We followed the procedure suggested by the author of the program and as implemented by others (Jombart 2008; Jombart et al. 2010; Vuillaume et al. 2015). Briefly, we first characterized the synthetic variables maximizing genetic variation in our samples (Principal Component Analysis). We then looked for the minimum number of clusters (K) best describing this variation using a K-means algorithm. We set the algorithm to investigate values of K ranging from 1 to 20, and we performed 1 × 107 iterations. Cluster membership was represented at the individual and population level using a bar-plot.
Captive genetic diversity
Once a reference database of the wild genetic variability was built, individuals of unknown wild origin, i.e., those collected from iguanarios, were tested for genetic variability in their nuclear and mitochondrial DNA. Many of the iguanarios we sampled were established more than 15 years ago with individuals likely brought from different areas across the DR. Since then haphazard mating is known to have occurred, as was conveyed in our informal discussion with iguanario staff. It is therefore probable that samples in this study may not have a wild genetic makeup, but rather a combination of genes from different populations. Nuclear markers are subject to recombination and can, therefore, demonstrate a composite genetic makeup over generations of admixture between isolated lineages. As mitochondrial DNA is maternally inherited, it doesn’t recombine, and can be helpful in understanding the origin of captive individuals given an existing pattern of mtDNA distribution in the wild (Gentile et al. 2013); however, it can also be misleading as only the maternal ancestry is represented. Therefore, we tried to infer the population of origin of the captive individuals by performing a population assignment test using a combination of mtDNA and nuclear markers and the Bayesian framework implemented in BAPS v6 (Corander et al. 2006, 2008). This approach has proven useful in other studies (Zarza et al. 2016). Briefly, we used the trained clustering algorithm implemented in the software, which allows the assignment of samples of unknown origin to predefined clusters. We combined nuclear and mtDNA data, with each mtDNA haplotype coded as diploid with a missing allele. We ran two independent analyses: first, we forced the software to assign the iguanas collected in captivity to any of the 14 wild sampled locations; second, we reran the analysis without any constraint, i.e., allowing the software to cluster the captive individuals in as many clusters as needed.
Results
Wild genetic diversity and distribution
We identified five unique mtDNA haplotypes in 120 wild individuals (Figs. 2, 4). No additional haplotypes were discovered in the 24 additional samples not included in further analysis. All individuals from the eastern region of the DR (sampling areas 11–13) and Mona Island (sampling area 14) shared the same unique haplotype (Hap. A, Figs. 2, 4). Individuals from the southwestern DR (sampling areas 1–5) were monomorphic for haplotype D (Figs. 2, 4). Samples collected from the region south of Enriquillo Lake (sampling area 6) all showed a distinct haplotype (Hap. E, Figs. 2, 4). Individuals from sampling areas 8–10 were monomorphic for haplotype C with the exception of one individual with haplotype D from sampling area 8 (Figs. 2, 4). Samples collected from Cabritos Island (sampling area 7) were characterized by the presence of haplotypes C and D, as well as haplotype B, not found elsewhere in the DR (Figs. 2, 4). We ran an AMOVA separating individuals into an eastern (sampling areas 11–14) and western (sampling areas 1–10) clade. This analysis was significant at all hierarchical levels investigated (Table 3). In particular, partitioning individuals into an eastern versus a western clade was sufficient to explain more than half of the genetic variance found in our samples (φCT = 0.520, Table 3). Haplotype D was the most divergent from all other haplotypes found in this study (Fig. 5) and its distribution corresponds closely to what was the southern paleo-island. Therefore, we ran an additional AMOVA clustering individuals from sampling areas 1 through 4, all fixed for haplotype D, in a separate group. This AMOVA was able to explain almost 70% of the genetic variance found on the island (φCT = 0.687, Table 3).

Distribution of genetic variation in mitochondrial and nuclear DNA markers from 120 wild iguanas across 14 sampling sites identified in Figs. 1 and 2. Each bar represents an individual. Top: distribution of the 5 different mtDNA haplotypes identified in this study. Middle: results of the STRUCTURE algorithm on microsatellite data. Bottom: results of the DAPC clustering algorithm on microsatellite data

Median Joining Network built with the five different haplotypes identified in this study. Colors coding is identical to Figs. 2 and 3. Pink circles represent haplotypes inferred by the median joining algorithm. Bars across the branches connecting the various haplotypes represent the number of nucleotide differences between any of the two considered haplotypes. The size of each circle is proportional to the haplotype’s frequency
None of the 18 microsatellite loci selected for our analysis showed evidence of null alleles with a frequency higher than 0.2 (Table 2). We therefore retained all markers for downstream analyses. Individuals collected from sampling areas three and four showed a slight but significant excess of homozygosity (Table 1). Pairwise FST comparisons between groups ranged from 0.079 (Group 1 VS Group 4) to 0.526 (Group 7 VS Group 10; Fig. 6; see SM Tables 1, 2 for all FST pairwise comparison values and their statistical significance). None of the analyzed loci showed evidence of significant departure from a model of neutral evolution (SM Fig. 1). Although we feel confident in our conclusions, we recognize the potential bias of interpreting FST values obtained by pairwise comparisons of sites with uneven and small sample size. Unfortunately, sampling wild C. cornuta in the DR can be extremely difficult given their rarity and elusive nature in some regions.

Heat map of pairwise FST estimated using microsatellite data between all groups of wild sampled sites for Cyclura cornuta in the Dominican Republic. All pairwise comparisons are significant at 0.05
The STRUCTURE analysis partitioned wild individuals in 10 different genetic clusters (Fig. 4, see also SM Figs. 2 and 3). Individuals from sampling area 14 clustered together, in a separate group. The individuals from the eastern DR (sampling areas 11–13) clustered together (Fig. 4). The majority of the other individuals were clustered in different groups corresponding to their sampling areas. We ran two separate AMOVAs, first partitioning individuals into western and eastern clades, where 16.8% of the genetic variance was explained by the eastern versus western separation (ϕCT = 0.168, Table 4). The second AMOVA partitioned the regions in the southern paleo-Island, northern paleo-Island, and western region of the DR, and was able to explain 11.4% of the genetic variance found (ϕCT = 0.114, Table 4).
The DAPC output produced a very similar pattern to that of STRUCTURE, but identified only seven clusters. Mona Island and the sampling areas from the eastern DR (areas 11–13) were again recognized as separate groups (Figs. 3, 4). Almost all individuals from sampling areas 1, 3–6, and 8 formed a single genotypic group. Samples from sites 2, 7, 9, and 10 were assigned to their own separate clusters, with the exception of a few individuals from sampling area seven who showed alternative group assignments (Figs. 3, 4).
Captive genetic diversity
No novel mtDNA haplotypes were discovered in the 114 captive samples analyzed (Fig. 7). Despite the large number of iguanarios located in the eastern DR, only three individuals had the wild haplotype from that area (i.e. haplotype A, Figs. 1, 2, 7), which is consistent with information gained from discussions with iguanario staff suggesting that most captive individuals originally came from the western DR, where hunting is most common. The analysis with BAPS shows that individuals sampled from captive facilities are likely the result of intraspecific hybridization events of parents coming from different areas across the country. Results from the BAPS constrained analysis grouped the majority of individuals with the wild individuals collected from sampling area three (SM Tab. 3). A few individuals were clustered with samples collected from area 5. A single captive sample from iguanario “Jurassic Park” was clustered with the only wild individual sampled from area 11, while the only three captive individuals with haplotype A were clustered with the wild individuals from sampling area 12, in the eastern region of the island (SM Tab. 3). When we ran the same analysis without constraining the captive individuals within any of the wild populations the results changed dramatically (SM Tab. 3), and all individuals were grouped in clusters inferred by the algorithm, suggesting that extensive recombination is occurring within the iguanarios.

Distribution of genetic variation in mtDNA from 114 iguanas sampled across different iguanarios (see Fig. 1 for location of various iguanarios). Each bar represents an individual. White bars indicate that no mtDNA sequence was obtained from those individuals
Discussion
Understanding the population genetic structure of threatened species is vital to developing appropriate management strategies, particularly when a metapopulation approach may be necessary due to a disjunct distribution (Akçakaya et al. 2007). We investigated the population genetic structure of Cyclura cornuta iguanas from most known regions of occurrence, fragmented across the Dominican Republic. Unfortunately, our data do not encompass the overall genetic diversity of the species, as we were unable to collect samples from Haiti. Further, due to the rare and elusive nature of this species in some areas of its range in the DR our sample sizes are sometimes low. Nonetheless, our results speak to the conservation of this species as we were able to: (1) identify patterns of significant population structure across the DR; (2) elucidate the possible detrimental effect that unmanaged captive facilities could have on wild iguana populations; and (3) shed light on the taxonomic dispute of Cyclura iguanas on Mona Island.
The collision of the north and south paleo-islands, resulting in the creation of Hispaniola, in the mid-Miocene, as well as subsequent sea inundations submerging the Enriquillo Basin until the mid-Pleistocene have impacted the genetic structure of many species (Glor et al. 2004; Townsend et al. 2007; Gifford and Larson 2008; Sly et al. 2011; Brace et al. 2012; Grigorev et al. 2018) (Fig. 1). A single C. cornuta mtDNA haplotype was found in the region corresponding to the southern paleo-island, or the area south of the Enriquillo Basin (haplotype D; Figs. 2, 4). The pattern found in the nucDNA is less clear, but a southern signature also exists (Fig. 3). This north–south pattern accounts for almost 70% of the total genetic variance found in the mitochondrial DNA (ϕCT = 0.687, Table 3) and 11.4% of the nuclear genetic variance (FCT = 0.114, Table 4). The southwest portion of the DR is the only sampled area in this study that occurs on the southern paleo-island, south of the Enriquillo Basin. Additional sampling along the Tiburon peninsula in Haiti will further elucidate the impact of the Enriquillo Basin in shaping the genetic structure of this species. Understanding which events, paleo-island collision or various sea inundations, caused the observed pattern is a more complicated issue as it depends upon an accurate divergence time estimate. Malone et al. (2017) estimated that the Cyclura clade dates to approximately 20 mya. Further, Reynolds (in prep.) dates the divergence of C. cornuta at approximately 2.5 mya based on a 50 PIC Bayesian BEAST analysis of Cyclura. This timing would reject the hypothesis that paleo-island collision and orogenic activity in the Miocene caused the divergences among populations of C. cornuta. Instead, a more likely scenario is that ancestral populations of C. cornuta colonized Hispaniola during sea level recession at the Pliocene/Pleistocene boundary, dispersing throughout the low-lying areas, and then becoming fragmented by sea inundations during Pleistocene interglacial periods (1–1.5 mya) (Raymo et al. 2006). More recent habitat degradation and hunting pressures have also likely impacted the current distribution, but on a finer scale. Further, the potential that human mediated movements have impacted the observed genetic structure must be considered. One such case, undertaken by the DR Ministry of Environment, is known (Secretaria de Estado de Medio Ambiente y Recursos Naturales 2001; Secretaria de Estado de Medio Ambiente y Recursos Naturales 2002); however, the relocation was not far from the point of origin. Another such case has been mentioned for Cabritos Island; however, there are no written accounts of this movement. In general, human mediated movements are rare, usually involving few individuals, and short distances from the origin. Thus, the impact of these movements on the observed genetic structure is likely minimal.
Both mitochondrial and nuclear genetic data reveal an elevated level of genetic variation within the Enriquillo Basin. Haplotype D is found within all individuals sampled from the area south of the Enriquillo Basin, as well as within the Enriquillo Basin (Figs. 2, 4). Likewise, individuals sampled from the south-central and northwestern populations in the DR (areas 9 and 10) share the same mtDNA (haplotype C; Figs. 2, 4), which can also be found in two of the sampling areas within the Enriquillo Basin. Additionally, two unique mtDNA haplotypes (B and E) were discovered within two sampling regions (6 and 7) in the Enriquillo Basin (Figs. 2, 4). DAPC results also indicate a common genetic signature across the southern sampling locations, which extends into the Enriquillo Basin, and a shared genetic signature between sampling area 10, in the south-central region, and the Enriquillo Basin (Figs. 3, 4). Further, there is novel variation found within the basin (Figs. 3, 4). The STRUCTURE results exhibit a similar pattern but further divide the areas in the western DR, such as separating sampling areas 5 and 6 in the southern portion of the Enriquillo Basin (Fig. 4). This is likely indicative of sea inundation, followed by secondary colonization into this region following sea level recession in the Pleistocene. This increased genetic variation demonstrates that this region is of great conservation importance when considering the protection of diversity within this species.
The Dominican Republic has a complex system of mountains, holding the record for the highest mountain peak among all Caribbean islands. These mountains, which stabilized in the mid-Miocene, continue to inhibit migration for species preferring xeric lowlands. Pico Duarte, reaching 3098 m a.s.l., is found within the Cordillera Central, or the central mountain range, of Hispaniola (Fig. 1; Nairn and Stehli 1975). This mountain range represents a natural barrier dividing the island from the south-central region of the DR to the northwest of Haiti. Wild Cyclura cornuta samples collected east of this mountain range all presented a distinctive and unique mtDNA haplotype (Hap. A), and nuclear genotypes separate them from iguanas sampled throughout the rest of the DR (Figs. 2, 3, 4). This east–west pattern accounts for over 50% of the total genetic variance found at the mitochondrial DNA level (ϕCT = 0.520, p value < 0.001), and 15% of the variance found at the nuclear DNA level (FCT = 0.154, p value < < 0.001). Individuals collected from Mona Island fall within this eastern group based on their mitochondrial DNA (Figs. 2, 4). However, nuclear genetic variation further partitions the eastern group, with individuals from Mona Island recognized as a separate cluster (Figs. 3, 4; see below for a more detailed discussion).
Although areas 9 (group 7) and 10 (group 8) share a mtDNA haplotype, the nuclear data differentiates these areas (FST = 0.325, p value < < 0.000; Figs. 3, 4, 6). Further, area 9 (group 7) is one of the most genetically distinct locations within the DR (FST values ranging from 0.290 to 0.479) (Fig. 6; see SM Tabs. 1, 2). This is consistent with the Cordillera Central mountain region, considerable geographic distance separating these two locations, and sea inundations during interglacial periods likely isolating these regions from all other locations (Schubert 1984; Iturralde-Vinent and MacPhee 1999). Lastly, there is a novel nuclear genetic signature in sampling area 2, which is consistent with this location being a small cay (Figs. 3, 4). The multifaceted geologic history of Hispaniola, coupled with prominent geographic barriers, have contributed to the complex pattern of genetic differentiation seen today.
The genetic variability and distribution in C. cornuta present important issues related to the conservation and management of this species. In our analysis, we only recovered five mtDNA haplotypes from over 250 sequenced individuals, with some of them restricted to small geographic areas. Although there may be additional variation in Haiti, our sampling encompasses most known areas in the DR where wild populations of C. cornuta are known to persist (Glor et al. 2000). The total surface area of the DR is over 48,000 km2. The distribution of Rhinoceros Rock Iguanas is much smaller, estimated is to be approximately 10,000 km2 (Pasachnik and Carreras-De León 2019). Finding only five mitochondrial haplotypes in a species with a wide-spread distribution is unexpected. For comparison, the Critically Endangered C. carinata in the Turks and Caicos Islands, is known to persist on a total surface area no larger than 13 km2 (Gerber 2004). A recent study on the genetic structure of this species identified six different mtDNA haplotypes, using only a similar section of the ND4 region (Welch et al. 2017). Low levels of genetic variability at the mitochondrial level have been associated with selective sweep events, fixing one or few haplotypes (Ballard and Whitlock 2004), or with a dramatic reduction in population size. Based on the analysis of microsatellites, we did not find any evidence of significant departure from a model of neutral evolution at any site (SM Fig. 1). Thus, the latter hypothesis seems more plausible here, although selective sweep events may have occurred and should be evaluated with additional genetic markers. Pasachnik and Carreras De León (2014) report that recent surveys in areas with previously documented iguana populations have resulted in no sightings or very scarce signs of iguanas. This suggests that the conservation status of C. cornuta is more grave than previously thought, and an updated IUCN Red List Assessment now considers the species Endangered (Pasachnik and Carreras De León 2019). Future work using morphological characters and genetic data with higher resolution should focus on elucidating adaptive differences between populations across its patchy distribution.
Information collected from wild iguanas provides a baseline population structure that can be used to compare captive individuals and determine their origin. Mitochondrial DNA data can, at times, be sufficient to determine the general source location of an individual of uncertain origin (Gentile et al. 2013), especially if captured from the wild. The inclusion of nuclear DNA will; however, aid in these efforts, elucidating a more thorough genetic history of the individuals in question (Zarza et al. 2016). In this study, the analysis of mtDNA sequences from captive individuals did not reveal any novel haplotypes; however, our microsatellite data demonstrated that individuals did not genetically resemble those found in the wild. In particular, our results show that many of the captive facilities harbor individuals whose genetic composition is likely the result of interbreeding between geographically and genetically separated groups. Predicting the long-term evolutionary impacts of these breeding events between naturally segregated lineages is difficult. However, genetic data as well as information from discussions with staff suggests that the majority of individuals entering into captive facilities from the wild come from areas in the southwest. The majority of large-scale iguanarios occur in the southeast, thus the potential to negatively impact the wild populations in the southeast, which are genetically distinct, is probable. Given that iguanas have been shown to successfully mate with members of differentiated genetic clusters, species, and even other genera, these releases could have long-term negative impacts to the local populations (Pasachnik et al. 2009, 2011; Zarza et al. 2011, 2016) if local adaptations have evolved. Moreover, there are other threats to consider. The conditions within many of the captive facilities are poor, and diseases may be prevalent (Pasachnik and Carreras De León 2014). Therefore, as a cautionary method, the release of individuals from iguanarios into the wild should immediately cease and reproduction should be prevented at this time.
Resolving the taxonomic status of the Mona Island Cyclura population will require additional morphological data as well as more in-depth analyses of genetics data. Although originally described as a species, Cyclura stejnegeri (Barbour and Noble 1916), most authors have followed Schwartz and Carey (1977) and recognized the Cyclura population on Mona Island and the extinct C. onchiopsis from Navassa Island as subspecies of Cyclura cornuta prior to 2000. Schwartz and Carey (1977) did; however, discuss a number of morphologically unique characters distinguishing the Mona Island population from C. cornuta found in the DR, and other authors have more recently recommended recognizing all three taxa as separate species (Glor et al. 2000; Powell 2000; Powell and Glor 2000; Henderson and Powel 2009). Although the shared mtDNA haplotype between Mona and the eastern DR demonstrates common ancestry between these populations, pairwise FST comparisons using microsatellite data clearly indicate that the iguana population on Mona Island has been isolated for a significant amount of time (SM Tabs. 1 and 2). The geology of this area demonstrates that Mona Island was likely never connected to any other islands in its vicinity (Kaye 1959), suggesting that its flora and fauna are likely the result of various colonization events since its emergence in the Pliocene (Kaye 1959). Although it is difficult to be certain of the exact timing, it is clear that these populations have been reproductively isolated from one another for a substantial amount of time, gene flow is not occurring, and the Mona Island population is on its own evolutionary trajectory. We find there is potential for this population to be considered a distinct species, endemic to Mona Island; however, additional data are necessary to reach a robust conclusion.
Although additional genetic and morphological data are needed to elucidate adaptive differences across the known populations of Cyclura cornuta within the DR, our data demonstrate unique genetic signatures across the country, as well as shed light on the specific status of Cyclura iguanas on Mona Island. These results should be used to guide conservation actions and management decisions that preserve the greatest amount of genetic diversity across this region. Most importantly populations from the southwestern, northwestern, and eastern regions of the DR should be managed independently, and special attention should be given to the populations within the Enriquillo Basin as they harbor increased genetic diversity within a small area (see also Ar and haplotype diversity values from Tables 1, 3, 4). Further, unregulated reproduction within captive facilities should be terminated immediately to avoid further genetic admixture through breeding of individuals with different origins. Although wild C. cornuta populations are diminishing and becoming more threatened, iguanarios throughout the DR cannot be considered for conservation breeding and release at this time. The use of breeding and release for conservation should only be considered when an individual’s wild origin is known, no individuals in the program are the progeny of unregulated captive breeding, and a proper marking system and studbook for breeding can be created and managed. Plans are currently underway with a local non-governmental organization, Grupo Jaragua, and DR Ministry of the Environment to improve these facilities such that they can be considered for conservation and management purposes in the future.
References
Akçakaya HR, Mills G, Patrick DC (2007) The role of metapopulations in conservation. In: Macdonald DW, Service K (eds) Key topics in conservation biology. Blackwell Publishing, Oxford, pp 64–84
An J, Sommer JA, Shore GD et al (2004) Characterization of 20 microsatellite marker loci in the west Indian rock iguana (Cyclura nubila). Conserv Genet 5:121–125. https://doi.org/10.1023/B:COGE.0000014062.86556.e3
Avise JC (2005) Phylogenetic units and currencies above and below the species level. Phylogeny Conserv 76:101
Ballard JWO, Whitlock MC (2004) The incomplete natural history of mitochondria. Mol Ecol 13:729–744. https://doi.org/10.1046/j.1365-294X.2003.02063.x
Bandelt HJ, Forster P, Röhl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036
Barbour T, Noble GK (1916) A revision of the lizards of the genus Cyclura. Bull Mus Comp Zool 9:137–164
Brace S, Barnes I, Powell A et al (2012) Population history of the Hispaniolan hutia Plagiodontia aedium (Rodentia: Capromyidae): testing the model of ancient differentiation on a geotectonically complex Caribbean island. Mol Ecol 21:2239–2253. https://doi.org/10.1111/j.1365-294X.2012.05514.x
Carreras-De León R, Pasachnik SA, Gerber GP et al (2019) Genetic structure at three spatial scales is consistent with limited philopatry in Ricord’s Rock Iguanas (Cyclura ricordii). Ecol Evol 9:8331–8350. https://doi.org/10.1002/ece3.5414
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631. https://doi.org/10.1093/molbev/msl191
Colosimo G, Knapp CR, Wallace LE, Welch ME (2014) Inferred vs realized patterns of gene flow: an analysis of population structure in the Andros Island Rock Iguana. PLoS ONE 9:e106963. https://doi.org/10.1371/journal.pone.0106963
Corander J, Marttinen P, Mäntyniemi S (2006) A Bayesian method for identification of stock mixtures from molecular marker data. Fish Bull 104:550–558
Corander J, Marttinen P, Sirén J, Tang J (2008) Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinf 9:539. https://doi.org/10.1186/1471-2105-9-539
Dempster AP, Laird NM, Rubin DB (1977) Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B 39:1–38. https://doi.org/10.1111/j.2517-6161.1977.tb01600.x
Dray S, Dufour A-B (2007) The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. https://doi.org/10.18637/jss.v022.i04
Earl DA, VonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Edelist C, Lexer C, Dillmann C et al (2006) Microsatellite signature of ecological selection for salt tolerance in a wild sunflower hybrid species, Helianthus paradoxus. Mol Ecol 15:4623–4634. https://doi.org/10.1111/j.1365-294X.2006.03112.x
Ellegren H (2004) Microsatellites: Simple sequences with complex evolution. Nat Rev Genet 5:435–445. https://doi.org/10.1038/nrg1348
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491. https://doi.org/10.1007/s00424-009-0730-7
Excoffier L, Laval G, Schneider S, Lischer HEL (2010) Arlequin (version 3.5): an integrated software package for population genetics data analysis. Evol Bioinform Online 10:564–567
Falush D, Stephens M, Pritchard JK (2007) Inference of population structure using multilocus genotype data: dominant markers and null alleles. Mol Ecol Notes 7:574–578. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180:977–993. https://doi.org/10.1534/genetics.108.092221
Frankham R, Ballou JD, Briscoe DA (2010) Introduction to conservation genetics. Cambridge University Press, Cambridge
Gentile G, Ciambotta M, Tapia W (2013) Illegal wildlife trade in Galápagos: molecular tools help the taxonomic identification of confiscated iguanas and guide their rapid repatriation. Conserv Genet Resour 5:867–872. https://doi.org/10.1007/s12686-013-9915-7
Gerber GP (2004) Cyclura carinata. The IUCN Red list for threatened species. http://dx.doi.org/10.2305/IUCN.UK.2004.RLTS.T6026A12317199.en. Accessed 16 Oct 2015
Gifford ME, Larson A (2008) In situ genetic differentiation in a Hispaniolan lizard (Ameiva chrysolaema): a multilocus perspective. Mol Phylogenet Evol 49:277–291. https://doi.org/10.1016/j.ympev.2008.06.003
Gifford ME, Powell R, Larson A, Gutberlet RL (2004) Population structure and history of a phenotypically variable teiid lizard (Ameiva chrysolaema) from Hispaniola: the influence of a geologically complex island. Mol Phylogenet Evol 32:735–748. https://doi.org/10.1016/j.ympev.2004.04.003
Gilbert KJ, Andrew RL, Bock DG et al (2012) Recommendations for utilizing and reporting population genetic analyses: the reproducibility of genetic clustering using the program structure. Mol Ecol 21:4925–4930. https://doi.org/10.1111/j.1365-294X.2012.05754.x
Glor RE, Warren D (2011) Testing ecological explanations for biogeographic boundaries. Evolution (N Y) 65:673–683. https://doi.org/10.1111/j.1558-5646.2010.01177.x
Glor RE, Powell R, Parmerlee JS (2000) Cyclura cornuta (Bonaterre). Hispaniolan Rhinoceros Iguana. Cat Am Amphib Reptil 709:1–6
Glor RE, Gifford ME, Larson A, Losos JB, Schettino LR, Lara ARC, Jackman TR (2004) Partial island submerence and speciation in an adaptive radiation: a multilocus analysis of the Cuban green anoles. Proc R Soc Lond B 271:2257–2265
Goudet J (2005) HIERFSTAT, a package for R to compute and test hierarchical F-statistics. Mol Ecol Notes 5:184–186. https://doi.org/10.1111/j.1471-8278.2004.00828.x
Graham A (2003) Geohistory models and Cenozoic paleoenvironments of the Caribbean region. Syst Bot 28:378–386. https://doi.org/10.2307/3094007
Grant TD, Hudson RD (2015) West Indian iguana Cyclura spp reintroduction and recovery programmes: zoo support and involvement. Int Zoo Yearb 49:49–55. https://doi.org/10.1111/izy.12078
Grativol AD, Ballou JD, Fleischer RC (2001) Microsatellite variation within and among recently fragmented populations of the golden lion tamarin (Leontopithecus rosalia). Conserv Genet 2:1–9. https://doi.org/10.1023/A:1011543401239
Grigorev K, Kliver S, Dobrynin P, Komissarov A, Wolfsberger W, Krasheninnikova K, Afanador-Hernandez YM, Brandt AL, Paulino LA, Carreras R, Rodriguez LE, Nunez A, Brandt JR, Silva F, Hernandez-Martich JD, Majeske AJ, Antunes A, Roco AL, O'Brien SJ, Martinez-Cruzado JC, Oleksyk TK (2018) Innovative assembly strategy contributes to understanding the evolution and conservation genetics of the endangered Solenodon paradoxus from the island of Hispaniola. GigaScience 7:1–21
Henderson RW, Powel R (2009) Natural history of west Indian reptiles and Amphibians. University Press of Florida, Gainsville
Iturralde-Vinent M, MacPhee RD (1999) Paleogeography of the Caribbean Region: implications for Cenozoic biogeography. Bull Am Museum Nat Hist 238:1–95. https://doi.org/10.2747/0020-6814.48.9.791
ITWG (2016) A checklist of the iguanas of the world (Iguanidae; Iguaninae). In: Iverson JB, Grant TD, Knapp CR, Pasachnik SA (Eds.). Iguanas: biology, systematics, and conservation. Herpetol Conserv Biol 11(6):4–46
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. https://doi.org/10.1186/1471-2156-11-94
Kamvar ZN, Tabima JF, Grunwald N (2013) The poppr R package for genetic analysis of populations with mixed (clonal/sexual) reproduction. Phytopathology 103:70
Kaye CA (1959) Geology of Isla Mona and notes on the age of Mona Passage
Lau J, Alberts AC, Chemnick LG et al (2009) Isolation and characterization of 23 polymorphic microsatellite loci for a West Indian iguana (Cyclura pinguis) from the British Virgin Islands. Mol Ecol Resour 9:1412–1414. https://doi.org/10.1111/j.1755-0998.2009.02683.x
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Longmire J, Maltbie M, Baker R (1997) Use of lysis buffer in DNA isolation and its implication for museum collection. Occas Pap 163:1–7
Malone CL, Wheeler T, Taylor JF, Davis SK (2000) Phylogeography of the Caribbean rock iguana (Cyclura): implications for conservation and insights on the biogeographic history of the West Indies. Mol Phylogenet Evol 17:269–279. https://doi.org/10.1006/mpev.2000.0836
Malone CL, Reynoso VH, Buckley L (2017) Never judge an iguana by its spines: Systematics of the Yucatan spiny tailed iguana, Ctenosaura defensor (Cope, 1866). Mole Phyl Evol 115:27–39
Mann P, Draper G, Lewis JF (1991) An overview of the geologic and tectonic development of Hispaniola. Geol Soc Am Spec Pap 262:1–28. https://doi.org/10.1130/SPE262-p1
Maurasse FJ-M (1982) Survey of the geology of Haiti
Ministerio de Medio Ambiente y Recursos Naturales (2014) Quinto informe nacional de biodiversidad
Nairn AEM, Stehli FG (1975) The ocean basin and margins. The Gulf of Mexico and The Caribbean. Plenum Press, New York
Pasachnik SA, Carreras De León R (2014) Lost Iguanas: trouble in paradise. IRCF Reptil Amphib 21:1–8
Pasachnik SA, Carreras De León R (2019) Cyclura cornuta, The IUCN Red List of Threatened Species 2019: e.T6042A3099941. In: IUCN Red List Threat. Species. Doi: https://doi.org/10.2305/IUCN.UK.2019-%0D2.RLTS.T6042A3099941.en
Pasachnik SA, Fitzpatrick BM, Near TJ, Echternacht AC (2009) Gene flow between an endangered endemic iguana, and its wide spread relative, on the island of Utila, Honduras: when is hybridization a threat? Conserv Genet 10:1247–1254. https://doi.org/10.1007/s10592-008-9692-0
Pasachnik SA, Echternacht AC, Fitzpatrick BM (2011) Population genetics of the Honduran spiny-tailed iguana Ctenosaura melanosterna: Implications for conservation and management. Endanger Species Res 14:113–126. https://doi.org/10.3354/esr00342
Powell R (2000) Cyclura onchiopsis Cope. Navassa Island Rhinoceros Iguana. Cat Am Amphib Reptil 710:1–4
Powell R, Glor RE (2000) Cyclura stejnegeri Barbour and noble. Mona Island Rhinoceros Iguana. Cat Am Amphib Reptil 711:1–4
Powell R, Nieves DM, Gifford ME, Fontenot BE (2002) The “rhino factory” at Manati Park. Iguana Times 9:75–81
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Puechmaille SJ (2016) The program structure does not reliably recover the correct population structure when sampling is uneven: subsampling and new estimators alleviate the problem. Mol Ecol Resour 16:608–627. https://doi.org/10.1111/1755-0998.12512
R Development Core Team (2011) R: a language and environment for statistical computing. R Found Stat Comput 1:409
Ralls K, Ballou JD, Dudash MR et al (2017) Call for a paradigm shift in the genetic management of fragmented populations. Conserv Lett. https://doi.org/10.1111/conl.12412
Raymo ME, Lisiecki LE, Nisancioglu KH (2006) Plio-pleistocene ice volume, Antarctic climate, and the global 18O record. Science 313:492–495
Rodda GH, Bock BC, Burghardt GM, Rand AS (1988) Techniques for identifying individual lizards at a distance reveal influences of handling. Copeia 1988:905–913
Rosas KG, Pérez-Buitrago N, Acevedo JP et al (2008) Development and characterization of 11 microsatellite loci for the Mona Island iguana (Cyclura cornuta stejnegeri). Mol Ecol Resour 8:825–827. https://doi.org/10.1111/j.1755-0998.2007.02080.x
Rupp E, León YM (2009) Proyectos agrícolas al Sur del Lago Enriquillo : Amenaza seria para nuestra iguana de Ricord. Rev Atajo 8:16–17
Rupp E, León YM, Inchaustegui SJ, Arias Y (2009) Estudio de la tenencia de la tierra en el área de Cyclura ricordi en Pedernales. República Dominicana, Santo Domingo
Schubert C (1984) Investigation sobre el cuaternario de la Republica Dominicana. Revista Geografica 99:69–92
Schwartz A, Carey M (1977) Systematics and evolution in the West Indian iguanid genus Cyclura. Stud Fauna Curacao Caribb Islands 53:15–97
Searcy RA, Villers LM, Reams RD et al (2009) Captive reproduction of the Jamaican iguana (Cyclura collei). Zoo Biol 28:343–349. https://doi.org/10.1002/zoo.20236
Secretaria de Estado de Medio Ambiente y Recursos Naturales SD (2001) Memoria Año 2001
Secretaria de Estado de Medio Ambiente y Recursos Naturales SD (2002) Memoria Año 2002
Sly ND, Townsend AK, Rimmer CC et al (2011) Ancient islands and modern invasions: disparate phylogeographic histories among Hispaniola’s endemic birds. Mol Ecol 20:5012–5024. https://doi.org/10.1111/j.1365-294X.2011.05073.x
Taylor HR, Colbourne RM, Robertson HA et al (2017) Cryptic inbreeding depression in a growing population of a long-lived species. Mol Ecol 26:799–813. https://doi.org/10.1111/mec.13977
Townsend AK, Rimmer CC, Latta SC, Lovette IJ (2007) Ancient differentiation in the single-island avian radiation of endemic Hispaniolan chat-tanagers (Aves: Calyptophilus). Mol Ecol 16:3634–3642. https://doi.org/10.1111/j.1365-294X.2007.03422.x
Vuillaume B, Valette V, Lepais O et al (2015) Genetic evidence of hybridization between the endangered native species Iguana delicatissima and the invasive Iguana iguana (Reptilia, Iguanidae) in the Lesser Antilles: Management implications. PLoS ONE. https://doi.org/10.1371/journal.pone.0127575
Waples RS (1998) Evolutionarily significant units, distinct population segments, and the endangered species act: reply to Pennock and Dimmick. Conserv Biol 12:718–721. https://doi.org/10.1111/j.1523-1739.1998.97524.x
Welch ME, Colosimo G, Pasachnik SA et al (2017) Molecular variation and population structure in critically endangered Turks and Caicos Rock Iguanas: identifying intraspecific conservation units and revising subspecific taxonomy. Conserv Genet. https://doi.org/10.1007/s10592-016-0922-6
Welch ME, Long GJ, Berk JW et al (2011) Twenty-nine polymorphic microsatellite loci in Cyclura carinata, the Turks and Caicos Iguana, a critically endangered island endemic. Conserv Genet Resour 3:209–212. https://doi.org/10.1007/s12686-010-9324-0
Zarza E, Reynoso VH, Emerson BC (2011) Discordant patterns of geographic variation between mitochondrial and microsatellite markers in the Mexican black iguana (Ctenosaura pectinata) in a contact zone. J Biogeogr. https://doi.org/10.1111/j.1365-2699.2011.02485.x
Zarza E, Reynoso VH, Emerson BC (2016) Genetic tools for assisting sustainable management and conservation of the spiny tailed iguana Ctenosaura pectinata. Herpetol Conserv Biol 11:255–264. https://doi.org/10.1146/annurev.ecolsys.33.010802.150507
Acknowledgements
This study would not have been possible without financial support from the Institute for Conservation Research (ICR) at San Diego Zoo Global, the International Iguana Foundation, Mohamed bin Zayed Species Conservation Fund, Fondo Nacional de Innovación y Desarrollo Científico y Tecnológico (FONDOCyT) and Conservation International in providing financial support, and in-country support from Yolanda León, Ernst Rupp, and Sixto Incháustegui at Grupo Jaragua. SAP was supported by a postdoctoral research fellowship from the ICR during fieldwork. We would also like to thank José Luis Castillo, Gerson Feliz, Jairo Isaac Matos, Manuel Alejandro D Oleo, Jerbin Volquez, Anibal Volquez, Liz A. Paulino, Víctor I. De la Rosa, Víctor Hugo Reynoso, Benjamin Sawicki, Luis Ramirez, Jeffrey Corneil, Cristian Marte, and numerous local guides and iguanario caretakers for assistance in sample collection. This project was conducted under permits issued by the Ministerio de Medio Ambiente y Recursos Naturales of the Dominican Republic and IACUC approval from San Diego Zoo Global (project 12-003 and 14-037). Lastly, we would like to thank John Iverson and Kevin de Queiroz for their thoughtful reviews of this manuscript.
Funding
This study was funded by the Institute for Conservation Research (ICR) at San Diego Zoo Global, the International Iguana Foundation, Mohamed bin Zayed Species Conservation Fund, Fondo Nacional de Innovación y Desarrollo Científico y Tecnológico (FONDOCyT) and Conservation International.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Pasachnik, S.A., Colosimo, G., Carreras-De León, R. et al. Genetic structure of Rhinoceros Rock Iguanas, Cyclura cornuta, in the Dominican Republic, with insights into the impact of captive facilities and the taxonomic status of Cyclura on Mona Island. Conserv Genet 21, 837–851 (2020). https://doi.org/10.1007/s10592-020-01290-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-020-01290-6