
Abstract
Inbreeding poses a real or potential threat to nearly every species of conservation concern. Inbreeding leads to loss of diversity at the individual level, which can cause inbreeding depression, and at the population level, which can hinder ability to respond to a changing environment. In closed populations such as endangered species and ex situ breeding programs, some degree of inbreeding is inevitable. It is therefore vital to understand how different patterns of breeding and inbreeding can affect fitness in real animals. Domestic dogs provide an excellent model, showing dramatic variation in degree of inbreeding and in lifespan, an important aspect of fitness that is known to be impacted by inbreeding in other species. There is a strong negative correlation between body size and lifespan in dogs, but it is unknown whether the higher rate of aging in large dogs is due to body size per se or some other factor associated with large size. We used dense genome-wide SNP array data to calculate average inbreeding for over 100 dog breeds based on autozygous segment length and found that large breeds tend to have higher coefficients of inbreeding than small breeds. We then used data from the Veterinary medical Database and other published sources to estimate life expectancies for pure and mixed breed dogs. When controlling for size, variation in inbreeding was not associated with life expectancy across breeds. When comparing mixed versus purebred dogs, however, mixed breed dogs lived about 1.2 years longer on average than size-matched purebred dogs. Furthermore, individual pedigree coefficients of inbreeding and lifespans for over 9000 golden retrievers showed that inbreeding does negatively impact lifespan at the individual level. Registration data from the American Kennel Club suggest that the molecular inbreeding patterns observed in purebred dogs result from specific breeding practices and/or founder effects and not the current population size. Our results suggest that recent inbreeding, as reflected in variation within a breed, is more likely to affect fitness than historic inbreeding, as reflected in variation among breeds. Our results also indicate that occasional outcrosses, as in mixed breed dogs, can have a substantial positive effect on fitness.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Domestic dogs are one of the most numerous carnivores on the planet, but several features of their population structure make them a valuable model for conservation genomics and the management of small populations (Daniels and Bekoff 1989; Young et al. 2011). Despite the large total dog population, population sizes for purebred dogs are often small, particularly when considering the breeding population. Reproduction within breeds is usually tightly controlled, sometimes with the goal of modifying specific physical or behavioral traits, and sometimes with the goal of improving health and genetic diversity. There are also large populations of free-breeding dogs around the world, including stray/feral dogs and village dogs (i.e. dogs descended from historically free-breeding populations, in contrast to dogs that are crosses of pure breeds) (Boyko et al. 2009). These contrasting breeding styles and variable genetic structure across levels of the global dog population provide excellent opportunities for investigation of the genomic and fitness consequences of different breeding systems. Here, we investigate the effects of breeding systems and genomic diversity on a well-known phenomenon of fitness relevance in dogs: the strong inverse relationship between body size and life expectancy.
Aging in dogs
The domestic dog is well known for its astonishing phenotypic diversity. In variation of body size, the dog far surpasses all other domestic animals, with at least a 40-fold difference between the largest and smallest breeds (Wayne and Ostrander 1999). In addition to this diversity of physical form, domestic dog breeds also vary widely in traits of translational value, including disease prevalence and lifespan (Fleming et al. 2011; Kraus et al. 2013).
This broad, naturally occurring diversity is one of several features that make domestic dogs an attractive model for translational research. Extensive reviews of the subject have been published elsewhere (e.g. Kaeberlein et al. 2016; Gilmore and Greer 2015), but can be briefly summarized as follows: the breed structure and availability of extensive pedigree records increase the power of genetic studies, and a shared environment with humans, access to advanced medical care, and closer genetic similarity to humans compared to traditional model organisms all contribute to the value of dogs in translational research.
Specifically, the dog is an ideal model for research in aging and longevity due to its greater than two-fold natural variation in lifespan and considerable variation among breeds in the risk of specific age-related diseases (Fleming et al. 2011). An immediate goal for developing the dog as a model of aging is to identify genetic and environmental factors related to aging in dogs (Kaeberlein et al. 2016). It is well established that body size is an important predictor of lifespan across dog breeds: small breeds live longer than large breeds (Galis et al. 2007; Greer et al. 2007; Fleming et al. 2011). A recent analysis found that this pattern results from an accelerated rate of aging in larger breeds compared with smaller breeds and not from an inherently higher age-independent risk of mortality (Kraus et al. 2013). The inverse relationship between body size and longevity in dogs is not unique—the same pattern is observed in horses (Brosnahan and Paradis 2003; Miller and Austad 2005), mice (Miller et al. 2002), and possibly to some degree in humans (Samaras et al. 2003). However, these within-species patterns run counter to the general pattern observed across mammalian taxa, where small species like rodents typically live only a few years but large species like whales and elephants can live several decades or more (Calder 1984; Peters 1986; Read and Harvey 1989).
Various mechanisms have been proposed to explain the reduced lifespans of larger individuals within species. One hypothesis is that larger individuals are more susceptible to cancer. Large animals grow rapidly during development to reach their adult sizes, and it has been suggested that this makes it more likely that skipping a regulatory checkpoint will cause unchecked growth and tumorigenesis. It is also statistically more likely for a mutation that will lead to cancer to arise in a larger animal because they have more cells (Peto 1977). To combat this process, very large species like whales and elephants must have highly efficient tumor suppression mechanisms (Miller and Austad 2005; Abegglen et al. 2015). Indeed, a survey of causes of death in dogs in the UK suggested that large-breed dogs are over-represented among those dying of cancer compared with small breeds (Michell 1999), and certain types of cancer (e.g. osteosarcoma) certainly are much more common in large breeds (Rowell et al. 2011). Nevertheless, analyses of individual breeds have shown that even after excluding individuals dying of cancer, life expectancy remains low in large dogs (Bernardi 1988; Saint Bernard Club of America 1993). Thus, other factors must also contribute to the increased rate of age-related health decline in these dogs.
Inbreeding in dogs
Many traits, including body size, are under direct selection in dogs. The impressive phenotypic diversity present in dogs has arisen from intensive artificial selection on desired physical and behavioral traits over the course of dog domestication, particularly during the diversification of dog breeds over the last two centuries (Larson et al. 2012).
Dog breeders (and other animal breeders) establish new breeds by selectively breeding individuals that display certain desired traits. The term “inbreeding” was coined from the Victorian practice of “breeding in” a new feature, such as a curled tail or a specific coat pattern, by repeatedly crossing only dogs with that phenotype—typically beginning with parent/offspring or sibling crosses (Darwin 1868). This breeding strategy enables dramatically modified phenotypes to become fixed in a population quickly. However, multiple generations of inbreeding for the purpose of fixing a specific trait can result in offspring that are almost entirely homozygous. Alleles and traits besides those under active selection often “hitchhike” to high frequency in the process. Hitchhiking may result from physical linkage between genomic regions controlling multiple traits (Fay and Wu 2000), or from pleiotropic effects of the trait under selection. For example, in Chinese shar-peis, the same hyaluronic acid synthase variant that produces the breed’s distinctive wrinkled skin is implicated in the etiology of familial shar-pei fever (Olsson et al. 2011). In free-breeding populations both of dogs and of non-domestic species, genomic inbreeding (i.e. increasing loss of genomic diversity at either the individual or the population level) can result from strong directional natural selection or from reduction in population size.
Population reductions, such as those that occur at the formation of new breeds in domestic species, or in natural populations facing anthropogenic or other threats, often result in substantial loss of diversity due to genetic drift. In domestic dogs, the population expands after initial breed formation and new mutations can theoretically increase diversity over time, but in most breeds effective population size (Ne) is kept relatively small by tightly controlled breeding within closed populations (Leroy 2011). Dogs that are not wanted for breeding purposes are commonly neutered, and breeding pairs are usually selected and controlled by humans, so that the reproductive population (Ne) is much smaller than the census population. Diversity is lost by drift over time, and the population becomes increasingly homozygous. Overall, this reproductive pattern in domestic dogs has resulted in a high potential for inbreeding depression, even in the absence of genetic forces like hitchhiking that can increase the frequency of deleterious variants. Indeed, one recent study found evidence that bottlenecks associated with domestication and breed formation have produced an increased load of deleterious variation in dogs (Marsden et al. 2016), suggesting that on the timescale of dog breeding, genetic purging of deleterious variants through inbreeding and selection has not occurred to a level needed to avoid inbreeding depression.
Inbreeding depression occurs when increased homozygosity causes decreased fitness, due to either unmasking of recessive deleterious variants or the presence of overdominant loci, where the heterozygote has the highest fitness value. Inbreeding depression has been detected in many species in the form of reduced fertility (e.g. sperm abnormalities, Fitzpatrick and Evans 2009) and increased frequency of congenital disease (Khlat and Khoury 1991). (See also Hedrick and Kalinowski 2000 and Leroy 2014 for reviews of studies of inbreeding depression in endangered species and livestock species, respectively.) We might also expect a decrease in mean lifespan, a key component of classical fitness due to its effect on potential lifetime reproductive output. Lifespan is a function of age-specific probability of survival, which in turn can be viewed as the net effect of countless aspects of an animal’s overall health that may or may not impact reproduction. It has long been known that inbreeding can have a negative impact on lifespan under certain conditions. Classic studies in fruit flies showed that when inbred lines were crossed, the F1 hybrids had increased longevity compared to either parent strain (Hyde 1913; Pearl et al. 1923; Hollingsworth and Smith 1955), and more recent studies in diverse species including butterflies (Van Oosterhout et al. 2000), cattle (Sewalem et al. 2006), and gazelle (Cassinello 2005) have found that inbred animals have shorter lifespans relative to outbred conspecifics.
In addition to their phenotypic variation, dog breeds vary substantially in levels of mean genomic inbreeding and frequency of deleterious alleles. This variation, along with ready availability of hybrids in the form of mixed-breed dogs, presents an opportunity to investigate the effects of inbreeding on lifespan at different scales: individual, breed, and inbreeding status (pure vs. mixed). We hypothesized that inbreeding depression may be responsible for some of the variation in lifespan observed in dogs. In the following analyses, we tested this hypothesis by investigating the relationships between inbreeding, body size, and lifespan both among breeds and within a breed in order to more fully understand the aging process in the domestic dog.
Methods
Body size and demographic data
Male breed-average body weights (in kg) were obtained from Hayward et al. (2016). To reduce bias toward small breeds, we log transformed the weights for our analyses. log10(male average weight) is reported in Table S1 (ESM1). Information on annual registrations from 2000 to 2015 in 101 breeds was provided by the American Kennel Club (AKC). Average annual registrations provided a proxy for each breed’s population size.
Inbreeding
SNP genotype data were used to calculate inbreeding coefficients based on total autozygous segment length (calculated from runs of homozygosity) for 4586 dogs from 117 breeds. Each breed was represented by at least five individuals. Dogs were genotyped on a semicustom Illumina array; for detailed methods, see Shannon et al. (2015). To identify autozygous segments, SNP data were phased in SHAPEIT (Delaneau et al. 2012) and converted to PLINK.ped/.map format. Homozygous segments of 1 cM or more were identified in GERMLINE (Gusev et al. 2009). A maximum allowable mismatch rate was set at one homozygous marker and two heterozygous markers for a slice to be considered part of a match, and slice size was set at 32 markers. Total autozygous segment length (in cM) across the 38 autosomes was then calculated for each individual. Relative coefficients of inbreeding (F) were calculated for each dog by dividing its total autozygous segment length by the maximum observed autozygous length, found in a gray wolf sample (not included in later analyses). Breed-average F values are reported in Table S1 (ESM1).
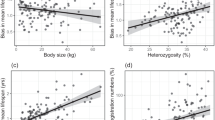
To increase the number of breeds in our analysis, we also used inbreeding estimates calculated from median heterozygosity (percent) scores provided by MyDogDNA® (Genoscoper Laboratories Oy), a commercial dog genetic testing service. Median heterozygosity was listed for 182 breeds with more than 30 individuals genotyped. Of those 182 breeds, 105 also had inbreeding estimates from autozygosity data. Heterozygosity and autozygosity were correlated in these 105 overlapping breeds, but two potential outliers (Studentized residual > |3|) were identified. Basenjis, which had a Studentized residual > |5| were excluded from further analysis. Bull terriers (Studentized residual = 3.57) were excluded from the regression model used to calculate adjusted coefficients of inbreeding, but were retained in later analyses due to their informative position near the upper end of observed inbreeding. With basenjis and bull terriers excluded, heterozygosity and autozygosity were correlated at a level of R2 = 0.65 (Fig. 1a).

Correlations and regression coefficients for adjustment of inbreeding and lifespan data from various sources. a Percent heterozygosity from MyDogDNA is negatively correlated with total autozygosity for 105 breeds in both data sets. Red points correspond to two outliers with Studentized residuals > |3| (BASJ = basenji, BTER = bull terrier). Linear regression parameters used to estimate inbreeding coefficients for breeds with heterozygosity data from MyDogDNA but no autozygosity data are given below the plot. b e(alpha) (life expectancy at onset of senescence) from Kraus et al. (2013) (K13) and median lifespan from Dobson (2013) (DOB) are both positively correlated with e(2) (life expectancy at age two) from the current analysis. Linear regression parameters used to estimate e(2) for breeds in Kraus et al. (2013) or Dobson (2013) but not the current analysis are given below the plot
Since both statistics are based on individual heterozygosity versus homozygosity, they can be readily compared. Total autozygosity (the percentage of the genome contained in long runs of homozygosity) should always be somewhat less than 1—% heterozygosity. After removing the outliers, we therefore used a linear regression model to compute estimated coefficients of inbreeding (F) for breeds with heterozygosity but no autozygosity data. An individual might have a higher or lower percentage of homozygous loci contained in runs of homozygosity than is typical for its breed, but when averaged over many dogs, regression should provide a good estimate of expected autozygosity at the breed level. Estimates were based on MyDogDNA heterozygosity values following the formula:
where “het” is median heterozygosity (on a scale of 0–1) from the MyDogDNA data and αhet is the intercept term from the regression model. Regression coefficients are given in Fig. 1a.
Generally speaking, total autozygosity is the definitive estimate of genomic inbreeding, since it is the most direct measurement of identity by descent. Calculating autozygosity requires either whole genome sequences or dense genome-wide marker data, which were not available from the MyDogDNA dataset. However, in many cases the MyDogDNA heterozygosity-based estimates were calculated from a greater number of individuals, so we had no clear a priori expectation of better breed-level accuracy for either estimate. Therefore, for breeds with data from both sources, we used an adjusted F value calculated as the average of F (observed based on autozygosity) and the F estimated from heterozygosity (expected autozygosity). For breeds present in only one dataset, we used whichever estimate of F was available. Bull terriers were included in both datasets, but since MyDogDNA heterozygosity was not well correlated with our autozygosity estimate (see above), we chose to use the estimate of F from our autozygosity data. Our sample size was fairly large for bull terriers (N = 39) and variance was low (σ2 = 0.008), so we believe our estimate is accurate. Adjusted inbreeding values are reported in Table S1 (ESM1).
In addition to genome-wide inbreeding metrics, we also calculated inbreeding estimates for specific regions of interest (methods in supplement, Figure S1, ESM2). Y-chromosome haplotype diversity provided a measure of inbreeding in the male line, while mitochondrial haplotype diversity revealed inbreeding in the female line. Nucleotide diversity (π) at major histocompatibility (MHC) loci provided estimates of diversity in a region that is known to be rapidly evolving and likely affects immunological fitness (Hughes and Yeager 1998).
Life expectancy
Data
Data on age at death and body size were extracted from the Veterinary Medical Database (VMDB http://www.vmdb.org/) spanning the years 1984 to 2004. The database provides information including breed (with mixed breed dogs comprising one category), age at death, and body size. Data on age at death and body size is provided in categories. For fitting mortality trajectories we used the following age at death categories: 2–4 years, 4–7 years, 7–10 years, 10–15 years, > 15 years. Because we focus here on adult lifespan, we only included dogs in the survival analysis that died after reaching their second year of life. In a previous study, we estimated that the onset of senescence, which was not significantly correlated with body size, averaged 2.15 ± 0.77 SD years for a set of 74 breeds, an age close to the left limit of the 2–4 year interval (Kraus et al. 2013). For comparing purebred and mixed breed dogs, we fitted the mortality functions separately for the following body size categories: in kg, midpoints 4.5, 10.2, 18.1, 28.4, 39.7, 56.7.
To exclude extrinsic causes of mortality, and because the prevalence of accidents has recently been shown to be higher in mixed breed dogs than in purebreds (Bellumori et al. 2013, also true for the VMDB Kraus et al., unpublished data), we excluded all deaths due to accidents including those due to intoxication [i.e. traumatic and toxic processes; for the classification of pathophysiological processes of causes of deaths see Fleming et al. (2011) and Hoffman et al. (2013)]. Results were qualitatively and quantitatively similar when using the full data set. Overall, our analyses of size-matched purebreds versus mixed breed dogs included 38,609 dogs (29,242 purebred and 9367 mixed breed dogs).
Survival analysis
Due to the categorical collection of ages-at-death we employed survival models for interval-censored data (Klein and Moeschberger 2003; Kraus et al. 2013). We fitted Gompertz mortality curves (hazard µ(age) = a*exp(b*age)) using maximum likelihood. We also tried other plausible parametric models such as the Gompertz-Makeham model or the three parameter Weibull model. However, each of these models led to convergence problems in some of the body size classes, probably due to the small number of intervals.
The target parameters we derived from our fitted functions were: (i) adult life expectancy, e(2 years), which we derived using numerical integration, (ii) the baseline hazard, i.e. the Gompertz parameter a which is the hazard level at the start of the mortality curve (initial mortality), (iii) the absolute rate of aging, i.e. the slope at ages 4, 7 and 10 years, and (iv) the relative rate of aging, i.e. the rate of aging relative to the current hazard, which in the Gompertz case is equal to the parameter b. See Kraus et al. (2013) for further details on these parameters. All estimates for model parameters are given in Tables S2 (across breeds) and S3 (purebred versus mixed breed dogs). Hazard curves are shown in Figure S3 (ESM2).
Due to idiosyncrasies of our data we have to acknowledge several assumptions and biases. Because we do not know the population at risk, we here have to assume that the analysed populations were stationary during the interval studied. Our models also assume that the age at death distribution is the same as in the population at large. This assumption is likely to be violated to some extent, because the cases dying at a veterinary teaching hospital are probably a non-random sample of diseases, ages and breeds. The comparatively low estimated values of life expectancy compared to other sources (e.g. Proschowsky et al. 2003; Michell 1999; O’Neill et al. 2013) might result from these violations. See Kraus et al. 2013 for further details.
Additional data sources
Longevity estimates for additional breeds were taken from Dobson (2013) and Kraus et al. (2013). The previous analysis by Kraus et al. used similar methods to the current paper except that dogs dying of as a result of accidents and those dying between 2 months and 1 year of age were kept in the analysis (see Kraus et al. 2013 for details). As a result, some breeds for which life expectancy could not be calculated in the current analysis due to insufficient sample size were included in that study. The current analysis therefore provides more accurate estimates of natural adult life expectancy (e(2)), but it is restricted to a smaller number of breeds. The Dobson (2013) analysis, meanwhile, reported median age at death based on results of a health survey of purebred dogs in the UK. This approach was less statistically rigorous, but nevertheless resulted in lifespan estimates that correlated well with our life expectancy estimates (Fig. 1b). In order to maximize power for our analysis, we therefore used linear regression models to compute adjusted e(2) values for those breeds present in Kraus et al. (2013) or Dobson (2013) but not the present analysis based on the formula:
where “e(alpha)” is life expectancy at onset of senescence from Kraus et al. (2013) and “med. lfsp.” Is median age at death from Dobson (2013). Regression coefficients are given in Fig. 1b. One potential outlier was identified based on Studentized residual > |3| (German shorthaired pointer, Studentized residual = 3.31), but due to negligible impact on model parameters, all breeds were retained in analyses. Longevity estimates are reported in Table S1 (ESM1).
Golden retriever data
To test the relationship between level of inbreeding and lifespan at the individual level, we used data from golden retrievers, collected by K9data.com, an open pedigree database. Individual lifespans and coefficients of inbreeding (CoI) were available for 9791 golden retrievers from around the world. CoI were based on pedigrees with variable numbers of generations and were expressed as percentages.
Statistical analyses
Statistical analyses were performed in R version 3.1.2 (R Core Team 2014). Linear models were used to test the relationships between adult life expectancy, body size, inbreeding, and population size. In the first model, autosomal inbreeding was the response variable, and log10(male average weight) and log10(average annual registrations) were the independent variables. In the second model, adult life expectancy (adjusted e(2)) was the response variable, and autosomal inbreeding coefficient (F) and log10(male average weight) (lbs) were the independent variables. For individual golden retriever data, linear regression was used to test the effects of sex and CoI (independent variables) on lifespan (response variable). T-tests were used to test for differences in lifespan between males and females and between inbred (CoI ≥ 2%) and individuals of each sex.
Results
Factors affecting inbreeding across breeds
Multiple regression analysis of the effects of body size and average annual registrations on autosomal inbreeding (F) shows that level of inbreeding is significantly related to body size, but not to the number of dogs registered (Fig. 2). Across 168 breeds, level of inbreeding tends to increase as body size increases (r = 0.18, p = 0.02). Regression coefficients are reported in Fig. 2c. Body size was more strongly correlated with inbreeding in the female line (mtDNA) than with inbreeding in the male line (chrY, Figure S1, ESM2).

Relationships between a census population size (N = 100 breeds) and b body size (N = 168 breeds) and breed-average level of inbreeding. Average annual number of AKC registrations for each breed is used as a proxy for population size. c Multiple regression on 100 breeds with complete data showed that number of registrations is not related to average coefficient of inbreeding, but body size is significantly positively related to inbreeding
Factors affecting life expectancy across breeds
Between purebred and mixed breed dogs, both purebred status and body size affected the key parameters of mortality trajectories (Table S3, ESM2). Compared to mixed breed dogs of a given size class, purebred dogs had a significantly lower adult life expectancy (Table 1, Fig. 3). On average, mixed breed dogs lived 1.2 years longer than purebred dogs. Together with body size, purebred status explained 95% of the variance in mean life expectancy, with purebred status alone explaining 46% of the variance. Purebred dogs had a significantly higher baseline hazard, with purebred status alone explaining 60% of the variance. Purebred dogs also had a significantly higher absolute rate of aging, which explained alone between 8% (at age 10) and 40% (at age 4) of the variance. Counterintuitively, the relative rate of aging was higher for mixed breed dogs, albeit not significantly different. Alone, purebred status explained 15% of the variance. As expected (Kraus et al. 2013), body size mainly affected life expectancy via the absolute rate of aging.

Life expectancy at 2 years of age is lower for purebred than for mixed breed dogs in each size class. Life expectancy estimates shown include the first 2 years of life
Across breeds of purebred dogs, adult life expectancy (adjusted e(2)) was negatively correlated with both body size (r = − 0.57) and coefficient of inbreeding (F) (r = − 0.14). Since body size and F were significantly positively correlated with each other, however, it was unclear whether the association with life expectancy was driven by body size, F, or both. Multiple regression analysis of the effects of body size and F on adult life expectancy showed that lifespan is highly significantly related to body size while variation in F had no additional effect (Fig. 4). This result supports the hypothesis that reduced lifespan in large breeds is mainly driven by their increased body size, and not by variation in the level of inbreeding between breeds.

Higher coefficients of inbreeding (F) are equally likely in breeds that are longer-lived or shorter-lived than expected based on their size. Life expectancy is plotted against log10(male average weight) for 105 breeds for which inbreeding estimates are also available. Trend line shows expected life expectancy based on regression on size only. Points are colored by breed-average level of inbreeding, such that outbred breeds are black and inbred breeds are red. Regression coefficients from regression of log10(male average weight) and coefficient of inbreeding (F) on adjusted life expectancy are given. Breed abbreviations are given in Table S4 (ESM2)
Factors affecting lifespan within a breed
Female golden retrievers tended to be longer-lived than males (Fig. 5a, t(9789) = 6.846, p = 8.03E−12), a trend that is observed in numerous other species as well (Austad and Fischer 2016). Within each sex, outbred individuals (CoI < 2%) tended to live longer than inbred individuals (Fig. 5b, c). Multiple regression of the effects of sex and inbreeding on lifespan showed that both sex and CoI have small but highly statistically significant effects on lifespan in golden retrievers (Fig. 5d). There was no significant interaction term between sex and CoI.

Density plots and regression coefficients showing the effects of sex and pedigree coefficient of inbreeding (CoI) in golden retrievers. a Female golden retrievers live slightly longer than males (t(9789) = 6.846, p = 8.03E−12), and outbred individuals (CoI < 2) live slightly longer than inbred individuals for both b) males (t(4305) = − 4.541, p = 5.74E−06) and c females (t(5482) = − 4.267, p = 2.02E−05). d Multiple regression confirms that being male and having a higher pedigree coefficient of inbreeding both negatively affect expected lifespan for golden retrievers
Discussion
Our results show that although large-breed dogs tend to have increased levels of inbreeding compared to small breeds, the reduced life expectancy of large dogs is driven by body size and not inbreeding depression. Furthermore, variation in inbreeding across dog breeds does not appear to be related to current breed population size. It should be noted, however, that census population size does not equal effective population size, and this inbreeding variation is likely related to breed differences in the strength of inbreeding at breed creation (founder effects) and/or differences in modern or historical breeding practices (e.g. use of popular sires). Our finding that body size and average level of inbreeding are positively correlated, particularly when considering inbreeding in the female line (Figure S1, ESM2), supports the conclusion that breeding practices do affect inbreeding variation across breeds. Specifically, since large breeds have larger litters (Borge et al. 2011), it is likely that fewer females reproduce in large breeds compared to small breeds, leading to reduced female effective population size and increased inbreeding. Sex-specific differences in reproductive behavior, such as polygyny in natural populations or the use of popular sires in domestic animal breeding, lead to differences in sex-specific effective population sizes. Our finding comes as both a contrast and a complement to past emphasis on popular sire effects in driving inbreeding-related problems in domestic animals (Leroy 2011). As dog breeders increasingly strive to establish breeding regimes that prioritize population health and sustainability, it will be important to consider female effective population size as well as limiting the use of popular sires.
Although inbreeding alone cannot explain patterns of deleterious genetic variation or inter-breed differences in fitness and longevity, this is not to say that it plays no role in the health or longevity of dogs. Indeed, our results both at the individual level and in comparing purebred versus mixed breed dogs indicate that inbreeding can have a highly significant effect on lifespan. Mixed breed dogs lived 1.2 years longer, on average, than size-matched purebred dogs (consistent with findings from Patronek et al. 1997), while outbred golden retrievers (CoI < 2%) were significantly longer-lived than inbred golden retrievers. Differences in genetic diversity at these levels (purebred status and within-breed variation) likely represent more recent inbreeding patterns, in comparison to historic differences that contribute to variation between breeds.
On the basis of these findings and population genetic theory, it would be reasonable to conclude that recent outcrossing produces hybrid vigor and positive effects on lifespan, while negative impacts on lifespan disproportionately result from recent inbreeding. The raw data we used to calculate inbreeding coefficients (autozygous segment lengths, identified from runs of homozygosity) allowed us to test whether breeds that are longer- or shorter-lived than expected based on their body size differ in burden of recent inbreeding. Autozygous segment length distribution provides information about the demographic history of a population—a large number of short autozygous segments suggests a small historic breeding population, while long segments are signatures of recent inbreeding. Contrary to our expectations, we found that there was no difference in recent inbreeding between dogs that are longer- or shorter-lived than expected for their size, but that shorter-lived dogs have somewhat stronger signatures of historic inbreeding than longer-lived dogs (Figure S4, ESM2, p = 0.12). This unexpected finding is readily explained, however, by the fact that nearly all of the dogs with large numbers of short autozygous segments belonged to brachycephalic breeds, which are likely to have reduced lifespans due to specific pathologies and respiratory constraints imposed by their skull morphology (Poncet et al. 2005; Bernaerts et al. 2010). Although the extreme brachycephaly that causes these pathologies is considered a recent development, selection for the general morphology extends back much farther, producing the historic inbreeding signatures we observed. Indeed, although the use of breed-average values seriously limits the power of genome wide association (GWA) studies in this case, the strongest association in a GWA analysis for life expectancy with size included as a covariate (Figure S5, methods in supplement, ESM2) was found in a SNP located within SMOC2, a previously identified candidate for effects on dog skull length (Boyko et al. 2010; Bannasch et al. 2010).
A recent study found that the increased genetic load in domestic dogs compared to gray wolves results from past bottlenecks, not recent inbreeding (Marsden et al. 2016). The authors of that study analyzed genome-wide patterns of deleterious variation and performed simulations to test whether various demographic models would produce the observed patterns. They found that even a high level of inbreeding in recent years (i.e. since the establishment of modern breeds) could not sufficiently explain the observed patterns of deleterious genetic variation without the effects of population bottlenecks at domestication and breed formation. Although we did not directly assess genetic load, our findings that (1) breed-average inbreeding is not related to current census population size and (2) traits with deleterious health effects such as brachycephaly are linked to historic inbreeding (i.e. from bottlenecks at breed formation), are consistent with the conclusions of Marsden et al.
Our tiered approach of examining effects of inbreeding on lifespan at three resolutions—within a breed, across breeds, and between mixed and purebred dogs—provides a system to detect interactions between evolutionary forces at work on different scales. Taken together, our results suggest that while some deleterious effects of old inbreeding from the formation of breeds are probably purged over time, outcrossing can yield substantial hybrid vigor, and further close inbreeding within a breed can still have negative impacts. Thus, it seems clear that individual-level phenotypes (lifespan and body size) from genotyped dogs would vastly improve power for detecting subtler effects of inbreeding, as well as specific genetic variants that may affect lifespan. For example, the genes in the IGF1 pathway are good candidates for explaining variation in longevity due to known involvement of IGF1 in body size in dogs (Eigenmann et al. 1984; Tryfonidou et al. 2003; Sutter et al. 2007) and lifespan in mice (Holzenberger et al. 2003). However, currently available data lacks sufficient resolution to detect effects of specific genotypes.
The domestic dog breed structure makes this model a powerful system for trait-mapping by GWAS (Sutter et al. 2004). When only breed-average phenotypes are available, however, this structure becomes a liability, since genetic differences between breeds reflect many phenotypic differences as well as neutral divergence. Currently, dates of death are available only for a small number of genotyped dogs (N = 38 at time of writing), but assuming pet owners remain willing to participate in research, more data will become available over time. In order to develop dogs as a translational model for aging research, future sampling efforts should therefore focus on obtaining individual-level phenotypes. Then, the combination of the breed structure and the powerful genomic tools available for the dog model may yield information on the genetic basis of aging not available from other systems.
Our conclusions in this study have direct applications for management of small populations to preserve genetic diversity and avoid inbreeding depression. These findings apply to managed breeding of endangered species (e.g. ex situ conservation programs) as well as the breeding of domestic animals, and can be summed up with the following:
-
1.
The reproductive output of individual females should be monitored and kept equal as much as possible to avoid loss of genetic diversity due to small female Ne, especially in species or breeds that produce large litters.
-
2.
Occasional crosses of animals from separate breeding pools (comparable to cross-breeding dogs or transfers of animals between separately managed international ex situ populations) can produce strong improvements in health and fitness.
-
3.
Unless detrimental traits are actively maintained in a population, fitness impacts of historical inbreeding may be mitigated by natural processes over time, as long as further close inbreeding is avoided.
References
Abegglen LM, Caulin AF, Chan A et al (2015) Potential mechanisms for cancer resistance in elephants and comparative cellular response to DNA damage in humans. JAMA 314:1. https://doi.org/10.1001/jama.2015.13134
Austad SN, Fischer KE (2016) Sex differences in lifespan. Cell Metab 23:1022–1033. https://doi.org/10.1016/j.cmet.2016.05.019
Bannasch D, Young A, Myers J et al (2010) Localization of canine brachycephaly using an across breed mapping approach. PLoS ONE 5:e9632. https://doi.org/10.1371/journal.pone.0009632
Bellumori TP, Famula TR, Bannasch DL et al (2013) Prevalence of inherited disorders among mixed-breed and purebred dogs: 27,254 cases (1995–2010). J Am Vet Med Assoc 242:1549–1555. https://doi.org/10.2460/javma.242.11.1549
Bernaerts F, Talavera J, Leemans J et al (2010) Description of original endoscopic findings and respiratory functional assessment using barometric whole-body plethysmography in dogs suffering from brachycephalic airway obstruction syndrome. Vet J 183:95–102. https://doi.org/10.1016/j.tvjl.2008.09.009
Bernardi G (1988) Longevity and morbidity in the Irish wolfhound in the United States—1966 to 1986. Harp Hound 1:78–84
Borge KS, Tønnessen R, Nødtvedt A, Indrebø A (2011) Litter size at birth in purebred dogs-A retrospective study of 224 breeds. Theriogenology 75:911–919. https://doi.org/10.1016/j.theriogenology.2010.10.034
Boyko AR, Boyko RH, Boyko CM et al (2009) Complex population structure in African village dogs and its implications for inferring dog domestication history. Proc Natl Acad Sci USA 106:13903–13908. https://doi.org/10.1073/pnas.0902129106
Boyko AR, Quignon P, Li L et al (2010) A simple genetic architecture underlies morphological variation in dogs. PLoS Biol 8:49–50. https://doi.org/10.1371/journal.pbio.1000451
Brosnahan MM, Paradis MR (2003) Demographic and clinical characteristics of geriatric horses: 467 cases (1989-1999). J Am Vet Med Assoc 223:93–98. https://doi.org/10.2460/javma.2003.223.93
Calder WA (1984) Size, function, and life history. Harvard University Press, Cambridge
Cassinello J (2005) Inbreeding depression on reproductive performance and survival in captive gazelles of great conservation value. Biol Conserv 122:453–464. https://doi.org/10.1016/j.biocon.2004.09.006
Daniels TJ, Bekoff M (1989) Population and social biology of free-ranging dogs, Canis familiaris. J Mammal 70:754–762
Darwin C (1868) The variation of animals and plants under domestication. D. Appleton, New York
Delaneau O, Marchini J, Zagury J-F (2012) A linear complexity phasing method for thousands of genomes. Nat Methods 9:179–181. https://doi.org/10.1038/nmeth.1785
Dobson JM (2013) Breed-predispositions to cancer in pedigree dogs. ISRN Vet Sci 2013:941275. https://doi.org/10.1155/2013/941275
Eigenmann JE, Patterson DF, Froesch ER (1984) Body size parallels insulin-like growth factor I levels but not growth hormone secretory capacity. Acta Endocrinol (Copenh) 106:448–453
Fay JC, Wu CI (2000) Hitchhiking under positive Darwinian selection. Genetics 155:1405–1413
Fitzpatrick JL, Evans JP (2009) Reduced heterozygosity impairs sperm quality in endangered mammals. Biol Lett 5:320–323. https://doi.org/10.1098/rsbl.2008.0734
Fleming JM, Creevy KE, Promislow DEL (2011) Mortality in North American dogs from 1984 to 2004: an investigation into age-, size-, and breed-related causes of death. J Vet Intern Med 25:187–198
Genoscoper Laboratories Oy MyDogDNA Technical Sheet–Design, Technology, and Performance Data
Galis F, Van Der Sluijs I, Van Dooren TJM et al (2007) Do large dogs die young? J Exp Zool B 308B:119–126. https://doi.org/10.1002/jez.b.21116
Gilmore KM, Greer KA (2015) Why is the dog an ideal model for aging research? Exp Gerontol 71:14–20. https://doi.org/10.1016/j.exger.2015.08.008
Greer KA, Canterberry SC, Murphy KE (2007) Statistical analysis regarding the effects of height and weight on life span of the domestic dog. Res Vet Sci 82:208–214. https://doi.org/10.1016/j.rvsc.2006.06.005
Gusev A, Lowe JK, Stoffel M et al (2009) Whole population, genome-wide mapping of hidden relatedness. Genome Res 19:318–326. https://doi.org/10.1101/gr.081398.108
Hayward J, Castelhano M, Oliveira K et al (2016) Complex disease and phenotype mapping in the domestic dog. Nat Commun. https://doi.org/10.1038/ncomms10460
Hedrick PW, Kalinowski ST (2000) Inbreeding depression in conservation biology. Annu Rev Ecol Syst 31:139–162
Hoffman JM, Creevy KE, Promislow DEL (2013) Reproductive capability is associated with lifespan and cause of death in companion dogs. PLoS ONE 8:e61082. https://doi.org/10.1371/journal.pone.0061082
Hollingsworth MJ, Smith JM (1955) The effects of inbreeding on rate of development and on fertility in Drosophila subobscura. J Genet 53:295–314. https://doi.org/10.1007/BF02993984
Holzenberger M, Dupont J, Ducos B et al (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421:182–187
Hughes AL, Yeager M (1998) Natural selection at major histocompatibility complex loci of vertebrates. Annu Rev Genet 32:415–435. https://doi.org/10.1146/annurev.genet.32.1.415
Hyde RR (1913) Inheritance of the length of life in Drosophila. Proc Indiana Acad Sci 23:113–123
Kaeberlein M, Creevy KE, Promislow DEL (2016) The dog aging project: translational geroscience in companion animals. Mamm Genome. https://doi.org/10.1007/s00335-016-9638-7
Khlat M, Khoury M (1991) Inbreeding and diseases: demographic, genetic, and epidemiologic perspectives. Epidemiol Rev 13:28–41
Klein JP, Moeschberger LP (2003) Survival analysis: statistical methods for censored and truncated data. Springer, New York
Kraus C, Pavard S, Promislow DEL (2013) The size-life span trade-off decomposed: why large dogs die young. Am Nat 181:492–505. https://doi.org/10.1086/669665
Larson G, Karlsson EK, Perri A et al (2012) Rethinking dog domestication by integrating genetics, archeology, and biogeography. Proc Natl Acad Sci USA 109:8878–8883. https://doi.org/10.1073/pnas.1203005109
Leroy G (2011) Genetic diversity, inbreeding and breeding practices in dogs: results from pedigree analyses. Vet J 189:177–182. https://doi.org/10.1016/j.tvjl.2011.06.016
Leroy G (2014) Inbreeding depression in livestock species: review and meta-analysis. Anim Genet 45:618–628. https://doi.org/10.1111/age.12178
Marsden CD, Ortega-Del Vecchyo D, O’Brien DP et al (2016) Bottlenecks and selective sweeps during domestication have increased deleterious genetic variation in dogs. Proc Natl Acad Sci 113:152–157. https://doi.org/10.1073/pnas.1512501113
Michell AR (1999) Longevity of British breeds of dog and its relationships with sex, size, cardiovascular variables and disease. Vet Rec 145:625–629. https://doi.org/10.1136/vr.145.22.625
Miller RA, Austad SN (2005) Growth and aging. Why do big dogs die young? Handb Biol Aging. https://doi.org/10.1016/B978-012088387-5/50022-4
Miller RA, Harper JM, Galecki A, Burke DT (2002) Big mice die young: early life body weight predicts longevity in genetically heterogeneous mice. Aging Cell 1:22–29
O’Neill DG, Church DB, McGreevy PD et al (2013) Longevity and mortality of owned dogs in England. Vet J 198:638–643. https://doi.org/10.1016/j.tvjl.2013.09.020
Olsson M, Meadows JRS, Truvé K et al (2011) A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet. https://doi.org/10.1371/journal.pgen.1001332
Patronek GJ, Waters DJ, Glickman LT (1997) Comparative longevity of pet dogs and humans: implications for gerontology research. J Gerontol A 52:B1717–B1718
Pearl R, Parker SL, Gonzalez BM (1923) Experimental studies on the duration of life. VII. The Mendelian inheritance of duration of life in crosses of wild type and quintuple stocks of Drosophila melanogaster. Am Nat 57:153–192
Peters RH (1986) The ecological implications of body size. Cambridge University Press, Cambridge
Peto R (1977) Epidemiology, multistage models, and short-term mutagenicity tests. Orig Hum Cancer 4:1403–1428
Poncet CM, Dupre GP, Freiche VG et al (2005) Prevalence of gastrointestinal tract lesions in 73 brachycephalic dogs with upper respiratory syndrome. J Small Anim Pract 46:273–279. https://doi.org/10.1111/j.1748-5827.2005.tb00320.x
Proschowsky HF, Rugbjerg H, Ersbùll AK (2003) Mortality of purebred and mixed-breed dogs in Denmark. Prev Vet Med. https://doi.org/10.1016/S0167-5877(03)00010-2
R Core Team (2014) R: a language and environment for statistical computing
Read AF, Harvey PH (1989) Life history differences among the eutherian radiations. J Zool 219:329–353. https://doi.org/10.1111/j.1469-7998.1989.tb02584.x
Rowell JL, McCarthy DO, Alvarez CE (2011) Dog models of naturally occurring cancer. Trends Mol Med 17:380–388. https://doi.org/10.1016/j.molmed.2011.02.004
Saint Bernard Club of America (1993) SBCA Health Survey. Saint Fancier 59–73
Samaras TT, Elrick H, Storms LH (2003) Is height related to longevity? Life Sci 72:1781–1802. https://doi.org/10.1016/S0024-3205(02)02503-1
Sewalem A, Kistemaker GJ, Miglior F, Van Doormaal BJ (2006) Analysis of inbreeding and its relationship with functional longevity in Canadian dairy cattle. J Dairy Sci 89:2210–2216. https://doi.org/10.3168/jds.S0022-0302(06)72291-3
Shannon LM, Boyko RH, Castelhano M et al (2015) Genetic structure in village dogs reveals a Central Asian domestication origin. Proc Natl Acad Sci 112:13639–13644. https://doi.org/10.1073/pnas.1516215112
Sutter NB, Eberle MA, Parker HG et al (2004) Extensive and breed-specific linkage disequilibrium in Canis familiaris extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. https://doi.org/10.1101/gr.3147604
Sutter NB, Bustamante CD, Chase K et al (2007) A single IGF1 allele is a major determinant of small size in dogs. Science (80-) 316:112–115
Tryfonidou MA, Holl MS, Vastenburg M et al (2003) Hormonal regulation of calcium homeostasis in two breeds of dogs during growth at different rates. J Anim Sci 81:1568–1580
Van Oosterhout CC, Zijlstra WG, van Heuven MK, Brakefield PM (2000) Inbreeding depression and genetic load in laboratory metapopulations of the butterfly Bicyclus anynana. Evolution (NY) 1:218–225. https://doi.org/10.1554/0014-3820(2000)054
Wayne RK, Ostrander EA (1999) Origin, genetic diversity, and genome structure of the domestic dog. BioEssays 21:247–257
Young JK, Olson KA, Sukh Amgalanbaatar RP, Berger J (2011) Is wildlife goint to the dogs? Impacts of feral and free-roaming dogs on wildlife populations. Bioscience 61:125–132. https://doi.org/10.1525/bio.2011.61.2.7
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Adam Boyko is the co-founder and Chief Science Officer of Embark Veterinary.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yordy, J., Kraus, C., Hayward, J.J. et al. Body size, inbreeding, and lifespan in domestic dogs. Conserv Genet 21, 137–148 (2020). https://doi.org/10.1007/s10592-019-01240-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01240-x