
Abstract
The endangered crocodile newt, Echinotriton andersoni, is a relatively large species of the family Salamandridae and is distributed on six islands in the central part of the Ryukyu Archipelago, Japan. Because of an originally small distribution range and recent habitat loss, this species has been steadily declining in number. To elucidate fine-scale population structure, which is essential for effective conservation management, we analyzed genetic diversity and gene flow based on nine microsatellite loci. Our results identified three different island groups (Amamioshima, Tokunoshima, and Okinawajima) and multiple genetic assemblages within the Amami and Okinawa island groups. The gross genetic variation within each island was positively correlated with island size. Population structure followed a latitudinal cline and isolation by distance, even among geographically isolated islands. In northern Okinawajima, relatively complex genetic structure was observed. This unexpected population structure seems to reflect historical migration and distribution expansion through the formation of land bridges and shifted coastlines in the Pleistocene. We also found that small islands showed little genetic variation (Ukeshima, Sesokojima, and Tokashikijima). In particular, our findings revealed that the Tokashikijima population is at greater risk for extinction than the other populations because it has the smallest effective population size.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genetic diversity is a key factor that ensures long-term persistence of a species and/or population in the face of changing environments because inbreeding and loss of genetic diversity reduce reproduction and survival in the short-term, and diminish the capacity of populations to evolve in response to environmental changes in the long-term (e.g., Frankham et al. 2002). Small populations of endangered species are more prone to inbreeding, which can further reduce genetic diversity and population sizes—a phenomenon known as the extinction vortex (Gilpin and Soulé 1986). The extinction vortex is especially common in small island populations because of their limited distributions and population size (e.g., Frankham 1997; Eldridge et al. 1999; Hinten et al. 2003; Lecis and Norris 2004; White and Searle 2007). Thus, understanding intra-island population structure, inter-island metapopulation structure, and mechanisms of population maintenance for a focal endangered island species is expected to be crucial for long-term sustainable management of species (Frankham et al. 2002).
The Ryukyu Archipelago is an island chain composed of approximately 140 subtropical islands that span approximately 1200 km and are located between Kyushu (Japan) and Taiwan. These islets are thought to have been connected by land bridges through diastrophism and sea level changes due to climatic change (e.g., Kizaki and Oshiro 1977, 1980; Osozawa et al. 2012). According to Kizaki and Oshiro (1977, 1980), Hikida and Ota (1997), and Osozawa et al. (2012), the Ryukyu region was located along the eastern coast of the Eurasian continent from the Miocene to the Pliocene, and submergence of the current Ryukyu Trough area and the two major straits (i.e., the Kerama Gap located between the Central and Southern Ryukyus and the Tokara Tectonic Strait between the Central and Northern Ryukyus) occurred during the Late Pliocene or Early Pleistocene. The complex origins of most islands in this archipelago and subsequent dynamic geographic change led to complicated dispersal—isolation events of terrestrial organisms, which enhanced their diversification and speciation. Consequently, this archipelago is characterized by high levels of species richness and endemism, despite its relatively small terrestrial area (e.g., Ikehara 1996; Ota 1998, 2000; Okamoto 2017). Because of their limited ability for overseas dispersal, 23 native amphibian species are distributed in this area, with high endemism in each island group. The endemism and extinction concerns are inextricably linked; 12 endemic amphibian species of the Ryukyus are listed in the IUCN Red List of Threatened Species (Kaneko and Matsui 2004).
Anderson’s crocodile newt (Echinotriton andersoni) is a relatively large newt that belongs to family Salamandridae; it is endemic to six small central Ryukyu islands: Amamioshima, Ukeshima, and Tokunoshima Islands of the Amami island group, and Okinawajima, Sesokojima, and Tokashikijima Islands of the Okinawa island group. In addition to having an intrinsically small, restricted habitat by the local paleoenvironmental changes (Nakamura and Ota 2015), recent habitat loss due to deforestation and development are causing population declines in E. andersoni. Therefore, E. andersoni has been listed as a class B1 endangered species in the IUCN Red List of Threatened Species (Kaneko and Matsui 2004). Hayashi et al. (1992) first reported relatively large genetic divergence between the Amami and Okinawa groups based on allozymes. Later, Honda et al. (2012) suggested a possible scenario of diversification across their current distribution based on analysis of the mitochondrial cytochrome b gene (cyt b); they found that E. andersoni haplotypes initially diverged between the two island groups during the Miocene before formation of the strait that then separated the two island groups. Moreover, several haplotype clusters diverged within the Okinawa island group. However, finer-scale population structure and demographic history within each island are still unknown. Some recent studies identified genetic differences among populations even on a small island (less than 100 km2) (White and Searle 2007; Igawa et al. 2013a; Wang et al. 2014). Specifically, Honda et al.’s (2012) analysis was based on a single mitochondrial gene, which is maternally inherited; thus, the fundamental population dynamics of this species are not fully understood.
For effective conservation management of endangered E. andersoni, it’s is important to elucidate fine-scale population structure and effective population size, especially based on nuclear genes. In particular the genetic information is essential for future reintroduction and genetic reinforcement for the small isolated populations. The objectives of our research were to investigate the population structure and demographic history of E. andersoni using microsatellite markers. We also examined patterns of gene flow that shaped population structures within each island, and discussed conservation concerns and sustainable conservation management of this species from a genetic perspective. Specifically, we discussed levels of genetic diversity on each island and correlation with island size and fine-scale population structures within each island and genetic diversity of local populations.
Materials and methods
Sampling, and genotyping
Between 2006 and 2012, we collected tissue samples of E. andersoni from 190 individuals from six islands by tail clipping (Fig. 1 and Table 1). Of these, samples from 81 individuals (12 from Amamioshima, three from Ukeshima, eight from Tokunoshima, 42 from Okinawajima, three from Sesokojima, and 13 from Tokashikijima) were the same as those previously used in Honda et al. (2012). Echinotriton andersoni is designated as a natural monument by Kagoshima and Okinawa Prefectural Governments, and its handling is thus regulated by law. Our sampling was conducted with permission from the Boards of Education of both governments. Additionally, the sampling was completed prior to designation of this newt as a rare species of wild fauna and flora of Japan by the national government in 2016.

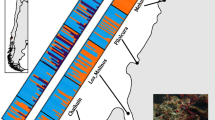
Mitochondrial genealogy, assigned genetic clusters, and sampling localities of E. andersoni. Tree: maximum likelihood tree based on mitochondrial cytochrome b haplotypes (1141 bp). This figure is adapted with permission from Honda et al. (2012)© (2012) Elsevier. Color scheme: graphical output of assigned genetic cluster from STRUCTURE (Hubisz et al. 2009). Each vertical bar represents an individual, and bars are divided into proportions of assigned genetic ancestral groups based on a the admixture model with the LOCPRIOR option and b the admixture model without any priors. Lines indicate the haplotypes of each individual determined by Honda et al. (2012). Map: sampling sites of individuals are indicated with solid circles on the shaded leaf maps of each island. Coloration of the circles represents assigned population membership from GENELAND (Guillot et al. 2005). Colored pie charts show the total ratios of assigned genetic ancestral groups of individuals in each population, as determined using STRUCTURE. Polygonal lines represent Voronoï tessellation calculated from BARRIER (Manni et al. 2004) and represent each population unit identified in GENELAND
Based on geographic location, islands except for Tokunoshima were divided into two groups: Amami island group (Amamioshima and Ukeshima) and Okinawa island group (Okinawajima, Sesokojima, and Tokashikijima). We tracked sample sites using a global positioning system in the field and treated the clusters of sites within an approximately 250-m radius as the same local population. Each population was labeled with a unique number (1–47). Although we tried to collect multiple samples from each locality, only a few individuals were collected in some localities because of low population densities and the endangered status of this species.
DNA was extracted from tissue using the DNeasy Tissue and Blood Kit (Qiagen, Hilden, Germany), and ten microsatellite loci (Sugawara et al. 2012) were examined. Loci were amplified using KOD FX (TOYOBO, Osaka, Japan) or ExTaq (TAKARA BIO, Otsu, Japan) following the protocol described in Igawa et al. (2011). Amplified fragments were electrophoresed on an ABI3130xl analyzer (Applied Biosystems), and allele sizes were determined using GeneScan LIZ 500 (Applied Biosystems) as an internal size standard and genotyped using GENEMAPPER 4.0 (Applied Biosystems). To test for repeatability in microsatellite scoring, we repeated all steps, from amplification through scoring, on a set of 48 samples. We used MICRO-CHECKER 2.2.3 (Van Oosterhout et al. 2004) to check for the presence of null alleles in local population from which at least ten individuals were collected (17, 25, 30, 31, 35, and 42). We also explored departure from Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) of the obtained loci using GENEPOP 4.0 (Rousset 2008) and GENALEX 6 (Peakall and Smouse 2006). The HWE and LD tests were assessed using the Markov chain algorithm (Guo and Thompson 1992).
Population structure analyses
We used an individual-based approach that does not assume existence of population in advance to explore fundamental population structure using Bayesian clustering analyses and tree clustering analysis based on proportion of shared alleles. We employed Bayesian clustering analyses implemented in STRUCTURE 2.3.3 (Hubisz et al. 2009) and GENELAND 4.0.3 (Guillot et al. 2005), both of which assign each individual to a genetic population unit. With STRUCTURE, we initially examined the entire dataset; then, we iteratively examined subdivisions of the dataset based on the island groups identified in the initial analysis. We conducted runs for which the number of clusters (K) varied from 1 to 10 for all individuals, and from 1 to 5 for each island group. We used admixture models for Markov Chain Monte Carlo (MCMC) inference with and without prior information on the locality of samples (LOCPRIOR). One million MCMC repetitions were conducted after discarding the first 100,000 iterations as burn-in. Ten simulations were completed for each estimated K. To estimate a realistic K value, we analyzed our results based on the methods described by Evanno et al. (2005), in which log-likelihood values and their variance from 10 repetitive runs for each K were used to calculate ΔK. We used the deltaK function of the CorrSieve package (Campana et al. 2011) in R 2.15.3 (R Core Team 2011) to calculate the ΔK. Replicate runs for each K were averaged using CLUMPP 1.1 (Jakobsson and Rosenberg 2007) and plotted using DISTRUCT 1.1 (Rosenberg 2004). The admixture coefficient (Q matrix) from STRUCTURE with the LOCPRIOR option for each island group was summed for each locality (Fig. 1). Spatial interpolation was also conducted to visualize the admixture coefficient in the Amami and Okinawa island groups using the krig function of the fields package in R.
For GENELAND, we separately conducted runs for each island group. Inferences were made using a spatial model without the ambiguity option, which assumes no-admixture and correlated allele frequencies. Lat/long of individual sampling sites were input as the spatial coordinates. We ran 10 independent runs, each of which consisted of 10,000,000 iterations with a thinning of 1000 after a burn-in of 2,500,000 iterations, and confirmed repeatability and convergence of the MCMC inference. Convergence was assessed by comparing the number of populations across replicate runs, with mean posterior density used as a criterion to choose the best run under a given set of model conditions. We treated geographically consecutive genetic clusters that were inferred in GENELAND as genetic population units for population-based analyses in the subsequent section, “Estimation of genetic diversity and demographic history.” Several genetic clusters that were spatially disrupted by other clusters were also treated as different population units (A6-1 and A6-2, and O1-1, O1-2 and O1-3).
For tree clustering analyses, we calculated proportion of shared alleles (Dps: Bowcock et al. 1994) between pairs of individuals using propShared of the adegenet package in R. Based on the distance matrix of Dps, we constructed a neighbor-joining tree (Saitou and Nei 1987) using the NEIGHBOR program in PHYLIP 3.695 (Felsenstein 1989). The resultant tree was visualized using TREEEXPLORE in MEGA 5.2 (Tamura et al. 2011).
Estimation of genetic diversity and demographic history
Observed and expected heterozygosities (HO and HE, respectively), observed number of alleles, effective number of alleles (AE), and fixation index (FIS) were calculated for each genetic population unit identified in GENELAND using GENALEX 6.5 (Peakall and Smouse 2006). Calculation of pairwise genetic distance, as determined based on FST and Nei’s DA (Nei et al. 1983), and construction of phylogenetic trees among genetic population units based on the DA with 1000 bootstrap iterations were separately performed using POPTREE2 (Takezaki et al. 2010) for each island group. We also visualized pairwise FST values between adjacent genetic population units that were higher than 0.1 using BARRIER 2.2 (Fig. 1) (Manni et al. 2004).
To test whether population structure was shaped by isolation by distance (IBD), we investigated correlation of Euclidean distance and the genetic distance matrixes using Mantel’s test (Mantel 1967). For each island group, we separately conducted distance matrix analyses for all localities except those with a sample size less than three. Linearized FST values (Rousset 1997) were used for genetic distances following the protocol described by Igawa et al. (2013a). The tests were conducted using FSTAT 2.9.3 (Goudet 2001). Correlation significance was determined through 2000 random matrix permutations.
Bottleneck detection
To detect molecular signatures of historical bottlenecks, we used two approaches that were implemented in BOTTLENECK 1.2.02 (Cornuet and Luikart 1996; Piry et al. 1999) and M-RATIO (Garza and Williamson 2001). BOTTLENECK investigates deviations from expected heterozygote excess relative to allelic diversity. During population bottlenecks, rare alleles are lost at a faster rate than loss of heterozygosity due to drift (Nei et al. 1975), and BOTTLENECK utilizes this disparity to detect past bottlenecks. We performed analyses under all three available microsatellite mutational models, infinite allele model (IAM), stepwise mutational model (SMM), and two-phase mutational model (TPM), with 80% single-step mutations and 20% multiple-step mutations. M-RATIO calculates the statistical significance of the M-statistic in each population compared with that of a simulated population (Garza and Williamson 2001). In this test, M is the ratio between the number of alleles at a given locus and the range of allele size. Because rare alleles are lost more regularly during a population bottleneck, M will be reduced in populations that have undergone a significant population size reduction, unless all rare alleles are within the predicted allele size distribution. We parameterized the TPM used in M-RATIO by assigning an 80% rate for single-step mutations, with a mean value of 2.8 repeats for the size change of multiple-step mutations, as previously discussed (Garza and Williamson 2001). Calculations were conducted under three θ (= 4Neµ) values: 0.05, 0.1, and 0.5. Because small sample sizes can skew results, we excluded genetic population units that contained fewer than five individuals (A1, A2, A4, A6-1, A6-2, A7, T4, O1-3, O4, and O5) from the analyses.
Results
Genotypic data
We excluded EanderPC-6 from the dataset because of low repeatability in some individuals. The total number of alleles ranged from 4 to 53, with a mean of 14.8 per locus. The mean number of alleles varied, with 8.0, 2.0, 7.4, 9.4, 1.8, and 1.7 alleles per locus in Amamioshima, Ukeshima, Tokunoshima, Okinawajima, Sesokojima, and Tokashikijima, respectively (Table S1). MICRO-CHECKER indicated the presence of null alleles at seven loci (EanderP-1, EanderM-1, EanderM-2, EanderPC-1, EanderPC-2, EanderPC-3, and EanderPC-4) in only one or two localities, and did not detect any evidence of large allele dropout or stuttering. HWE tests for all 54 polymorphic locus/population combination (α = 0.000926 after Bonferroni correction) were conducted and significant deviation from HWE (heterozygote deficiency) were detected in three tests of three loci (EanderPC-1 in 25, EanderPC-3 in 42, and EanderPC-4 in 25). No significant LD was detected (α = 0.000231 after Bonferroni correction). Because none of the loci showed significant HWE in most populations, none was excluded from analysis. We scored each individual twice to confirm consistent results between runs. Although we tried twice to amplify some loci of specific individuals, we could not unambiguously score 1.40% of genotypes, which were subsequently classified as missing data. Among islands, there were large differences in AE, HO, HE, and FIS (Table 1).
Population structure
Results of Bayesian clustering analyses using STRUCTURE are summarized in Fig. 2. In the analyses of the entire dataset that used an admixture model, the mean log posterior probability value, Ln P(D), was found to incrementally increase with increasing K, and reached the highest values under K = 5 (Fig. 2a). Although the highest ΔK value was observed when K = 2, and there was a very low Ln P(D) when K = 1, the peak was observed under K = 5. Because clear tripartition was observed at K = 3, which corresponded to the geographic location of the islands, we subdivided the dataset into three island group datasets for subsequent analyses: Okinawa (Okinawajima, Sesokojima, and Tokashikijima), Tokunoshima (Tokunoshima), and Amami (Amamioshima and Ukeshima). In the Okinawa group, incremental increase of Ln P(D) and peaking of ΔK at K = 3 were observed in both admixture models with and without LOCPRIOR (Fig. 2a). The general framework of clustering at K = 3 did not differ between the two models, which indicates subdivision of Tokashikijima individuals that shared common ancestry with southern Okinawajima individuals, and there was admixture of the two Okinawajima ancestral elements (Figs. 1, 2a). A similar pattern was also observed in the Amami group, which yielded the highest values of Ln PD (D) and ΔK at K = 3, in which there was subdivision of Ukeshima individuals and admixture of two ancestral elements in Amamioshima (Figs. 1, 2b). Moreover, the Tokunoshima group showed decrease of Ln P(D) in both models, which indicates no significant population structure in Tokunoshima (Figs. 1, 2c).

Summary of results of analyses using STRUCTURE 2.3.3 (Hubisz et al. 2009). Color scheme: graphical output of assigned genetic cluster from STRUCTURE in a total dataset, b Amami, c Toku, d Okinawa groups. Line graph: Plots of mean Ln P(D) (black lines) and ΔK (red lines) statistics against K in each dataset and model. (Color figure online)
Geospatial visualization of the ancestral elements using kriging interpolation in the Amami and Okinawa groups revealed the inferred core and extent of the ancestral elements (Fig. 3). In the Amami group, distribution of each ancestral element showed nonrandom spatial patterns. Although the ratio of ancestral elements for each individual do not entirely correspond to phylogenetic position of the mitochondrial haplotypes inferred in Honda et al. (2012), the overall spatial distribution of each ancestral element roughly overlapped with that of the proposed mitochondrial clades (Fig. 2a–c). In the Okinawa group, each ancestral element showed a clinal pattern along the major axis of the islands and corresponded to the spatial distribution of mitochondrial clades (clade 3–4 in Fig. 3d, clades 3–7, 3–5 and 3–6 in Fig. 3e, and clade 3–8 in Fig. 3f).

Spatial interpolation of proportions of genetic ancestries for three clusters (K = 3) detected by STRUCTURE across Amami (a, b, and c correspond to sky blue, blue, and purple in Fig. 1, respectively) and Okinawa (d, e, and f correspond to orange, green, and yellow in Fig. 1, respectively) island groups. Red dots indicate sample localities. Bars indicate genetic ancestral group probabilities. Dashed circles correspond to distribution of mitochondrial haplotypes revealed by nested clade phylogeographical analysis as the three-step clades (Fig. 1 in Honda et al. 2012). (Color figure online)
GENELAND results are summarized in Fig. S2. The highest mean posterior density was obtained for K =7, K = 5, and K = 10 in Amamioshima, Tokunoshima, and Okinawajima groups. The resultant populations are shown in Fig. 1. Individuals generally clustered according to geographic proximity, except for northern Okinawajima, which showed a somewhat complicated pattern because the spatial extent of O1 overlapped with other clusters. We thus treated each fragmented O1 subgroup as a separate genetic population unit (O1-1, O1-2, and O1-3). Similarly, we separated A6 into A6-1 and A6-2 because these two localities (Ukeshima and the southwestern tip of Amamioshima) are clearly separated by the sea strait.
Bottleneck detection
A Wilcoxon test implemented in BOTTLENECK revealed a significant excess of heterozygosity compared with the expected equilibrium (P < 0.05) in the following genetic population units: A1 and A5 (under all models), T1 (under the SMM model), O2 (under the IAM model), O3 (under the TPM and SMM models), and O6 (under the IAM model) (Table 3). M-RATIO (Garza and Williamson 2001) detected a significant population bottleneck signal in all populations except O1-1, O1-2, O8, and O9 based on θ values; T2 had θ = 0.1 and 0.5, and O6 had θ = 0.5 (Table 3).
Intra- and inter-population diversity
Intra- and inter-population unit genetic diversity results are summarized in Tables 2 and S2, respectively. The number of effective alleles ranged from 1.47 (O9) to 3.27 (A3), with a mean of 2.27. The expected heterozygosities ranged from 0.187 (O9) to 0.558 (A3), with a mean of 0.408. Neighbor-joining trees based on the DA distance between genetic population units for the Amami and Okinawa groups were consistent with the results from STRUCTURE and mitochondrial data (Honda et al. 2012) (Fig. S3), and showed that clusters were composed of genetic population units that shared the same ancestral groups inferred in the STRUCTURE analyses (Fig. 2). In the Amami group, two clusters that were composed of A1, A2, A5, and A7, and A4, A6-1, and A6-2 received high bootstrap support (> 50%). In the Okinawa group, three clusters that were composed of O1-1, O1-2, and O1-3; O4 and O5; and O8 and O9 were also supported. In the Tokunoshima group, a cluster composed of T2 and T3 was also supported (Fig. S3).
As shown in Fig. 4, linear correlation between linearized FST and Euclidean distance were observed in inter- and intra-island groups, except in the intra-Tokunoshima group. Regression lines drawn through the scatterplot of these two variables showed the steepest slope within the Amami group. Mantel’s test on these distance matrixes within the Amami and Okinawa groups indicated a high Mantel’s r value (r = 0.858 and 0.575 within Amami and Okinawa groups, respectively) and significant correlations (both P = 0.0005). However, within the Tokunoshima group, regression lines did not show positive linear relationships between these two variables (r = − 0.865), but there was no significant correlation (P = 0.133).

Relationships between linearized FST and Euclidean distances between populations
Discussion
Gross genetic diversity of island populations
The determinant factor for genetic diversity of island populations varies depending on the evolutionary histories of target species and geographic background of islands. Previous studies that investigated genetic diversity of island populations relative to geographic features of the islands showed positive correlation of genetic diversity with island size (Frankham 1996; White and Searle 2007) or negative correlation of genetic diversity with time since island isolation (Wang et al. 2014).
Theoretically, reduction of genetic variation in island populations is unavoidable over time through the processes of genetic drift and inbreeding because of their limited population sizes, which should be correlated with island size. In this study, the size of islands where we collected samples was roughly divided into two classes: larger islands, including Amamioshima, Tokunoshima, and Okinawajima, and smaller islands, including Ukeshima, Sesokojima, and Tokashikijima (Table 1 and Fig. S1). The larger islands have more basic genetic diversity reflecting large differences of population sizes in these islands (data not shown). In particular, the Tokashikijima population had the smallest values of all estimators except F, which indicates that this population has the smallest effective population size and is of greatest conservation concern. Amamioshima had the highest diversity of the larger islands; for the smaller islands, Ukeshima had a similar diversity to Sesokojima, whereas Tokashikijima had a lower diversity. These differences can be explained in the context of intra-island population structure and recent migration from neighboring islands, as discussed in the next section.
Evolutionary history and population structure
Our series of genetic clustering analyses using STRUCTURE with multiple K values for the total dataset revealed divergence and genetic relationships among island populations. In brief, our results showed clear genetic divergence of three major genetic groups and genetic structure within each island.
The clear dichotomy of clusters at K = 2 indicates that the populations in Tokunoshima are genetically closer to the Amami group than Okinawa group (Fig. 2a, K = 2), which is consistent with the mitochondrial genealogy (Fig. 2 in Honda et al. 2012). However, different from the mitochondrial data, the trichotomy at K = 3 supported a split in Tokunoshima and inclusion of Ukeshima in the Amami group (Fig. 2a, K = 3), and existence of three major genetic groups, Amami, Tokunoshima, and Okinawa groups. Formation of clusters within the Okinawa group at K = 4 and 5 indicates that the ancestral population of Tokashikijima and the southern Okinawajima population first diverged from a common ancestral population in the Okinawa group; subsequently, two different ancestral populations diverged.
Overall, these divergence patterns are consistent with that of a molecular phylogeny based on the mitochondrial cyt b (Fig. 3), excluding the sister relationship of Ukeshima and Amamioshima populations (Fig. 1). Considering the geographic distance between Ukeshima and Amamioshima, the mitochondrial genealogy unlikely reflects the true ancestral population differentiation because it may have been affected by incomplete lineage sorting and/or lack of lineage-specific mutation accumulation due to the lower mutation rate of mitochondrial genes compared with microsatellite loci. Such conflict between mitochondrial and nuclear genes was also found in a recent study describing new species and relationships within Echinotriton (Hou et al. 2014), which may have been caused by lack of phylogenetically informative sites among Echinotriton species in pro-opiomelanocortin (only four or three substitutions in 475 bp of pro-opiomelanocortin).
Based on the divergence times estimated in Honda et al. (2012), these divergences among the islands occurred prior to submergence of the land bridges, which occurred approximately 1.5 Ma (Osozawa et al. 2012, 2013): 3.1–4.8 Ma between Amami + Tokunoshima and Okinawa island groups; 0.6–1.1 Ma among Tokunoshima, Ukeshima, and Amamioshima populations; and 0.8–1.2 Ma between northern-central and southern Okinawajima + Tokashikijima (Honda et al. 2012). As discussed below, our data showed genetic divergences among populations even within each island. Thus, the ancestral E. andersoni populations may have been isolated from each other by environmental factors prior to isolation by sea strait formation.
Within each island group, our genetic cluster assignment revealed multiple ancestral elements and genetic population units. In the Amami group, three major ancestral groups were estimated and assigned to seven population units. For the genetic divergence of these population units, all FST values between adjacent population units were greater than 0.1 (Fig. 1), which indicates a longer history of isolation, even between the geographically close populations; this is congruent with the biased distribution of haplotypes of a mitochondrial gene (Fig. 1 in Honda et al. 2012). However, distribution of the ancestral groups showed a clinal pattern (Figs. 1, 3), which indicates existence of long-term gene flow among populations. This pattern is explained by the clear IBD pattern of population diversification, which showed linear relationships of migration rate (linearized FST) and geographic distance (Fig. 4). This clinal distribution pattern might reflect the limited locomotor ability of this newt and fixation of alleles through drift and selection in each population. In addition, IBD was observed even between currently isolated islands; thus, this diversification pattern probably accumulated slowly over time. In particular, although the major two ancestral groups in Amamioshima overlapped between the northwest and southeast (Fig. 3b, c), local population 13 in the southwestern tip of Amamioshima and local population 16 in Ukeshima shared a relatively recent common ancestor and belonged to the same genetic population unit (Fig. 1). This relationship indicates historical gene flow between these populations via formation of a super-island, which included Ukeshima, Amamioshima, Kakeromajima, and Yorojima Islands, and is similar to the diversification pattern of Cynops ensicauda in these islands during the last glacial maximum of the Pleistocene (ca. 0.015–0.020 Ma) (Tominaga et al. 2010).
In the Okinawa group, three ancestral groups were distributed along the major axis of the islands with a latitudinal cline pattern. However, genetic distances between adjacent population units were not as prominent as in the Amami group, manifesting a gently sloping regression line in migration rate and geographic distance plots (Fig. 4). These differences between Amamioshima and Okinawajima were also observed in Ishikawa’s frogs (Odorrana splendida and O. ishikawae, which are endemically distributed in Okinawajima and Amamioshima, respectively). According to Igawa et al. (2013a), O. splendida in Amamioshima showed multiple distinct populations, which reflects the complex topography of this island, whereas O. ishikawae in the northern mountainous area of Okinawajima showed no obvious population structure. Consequently, differences in topography and/or environmental factors related to the geography of these islands should also affect migration rate and structure of E. andersoni populations. Population structure in the Okinawa group slightly differed from that of the mitochondrial genealogy; the ancestral group was predominantly distributed in Tokashikijima (46 and 47 in Figs. 1, 2d) and was also located in the southern half of Okinawa. Additionally, almost all alleles found in the O9 population unit in Tokashikijima were common alleles shared with Okinawajima populations. A possible explanation for the discrepancy between microsatellite and mitochondrial data are that there is a higher probability of mitochondrial haplotype fixation due to four-fold lower effective population size of mitochondrial genes compared with nuclear loci.
In the northern mountainous area of Okinawajima, a comparatively complex structure manifesting two different overlapping ancestral groups (Figs. 1, 3) was detected. Because the latitudinal stepping-stone migration along the current topography could not explain such a complicated pattern, this pattern might reflect a recent migration corridor that was directly linked to the middle and northwestern parts of Okinawa. Recently, unexpected fine-scale subdivision of salamander populations was detected, highlighting the effect of landscape, which has been recognized as an important factor that shapes gene flow corridors (Giordano et al. 2007; Wang et al. 2009; Savage et al. 2010).
According to Kan (2014), the coastline of the western side of northern Okinawajima was shifted to the west, and the land area extended from the Motobu Peninsula during the last glacial maxima. The ancestral O2 and O6 populations might have experienced gene flow through this extended land area. Hence, the complex population structure in the northern Okinawa area may result from recent migration and persistence of genetically heterogeneous populations. In this case, the mountainous landscapes in this region potentially served as a barrier that prevented rapid admixture among these adjacent populations. However, the substantially larger effective population size of O2 indicates that admixture is ongoing to some extent.
In contrast with the Okinawa and Amami island groups, no significant genetic divergence was detected in the Tokunoshima group. Although we detected five population units, only a single ancestral group was estimated. As shown in migration rate and geographic distance plots (Fig. 4), the population units in Tokunoshima are geographically less than 20 km apart and do not display an IBD pattern. In addition, despite such small inter-population diversity, the genetic diversity within each population was comparatively higher than observed in the other islands, as mentioned in the previous section. These results indicate that Tokunoshima populations belong to a large single gene pool that was maintained by frequent migration between geographically close populations; this inference is also supported by a high population density of this species in Tokunoshima (Utsunomiya et al. 1978).
Bottleneck detection and conservation perspective
We found evidence of population bottlenecks in all populations under all models in M-RATIO, but in only two populations under all models in BOTTLENECK, with both populations being in the Amami group (Table 3); bottlenecks in two populations in M-RATIO but no populations in BOTTLENECK were found in the Tokunoshima group; and bottlenecks in three populations in M-RATIO and no populations in BOTTLENECK were detected under all mutation models in the Okinawa group (Table 3). These results are very similar to those from a similar analysis of Ishikawa’s frogs (Igawa et al. 2013a), wherein a genetic signature of bottleneck was revealed in the distant but not recent pasts, as BOTTLENECK is more effective for identifying populations that have recently experienced a severe reduction in population size. Alternatively, M-RATIO is likely to identify a bottleneck that occurred in the distant past, when reduction in population size was fairly severe, and also when the effective population size of the ancestral population was large. Our results indicated that bottleneck effects in Echinotriton newts on all islands were also substantial in the distant past; thus, recent concerns regarding population declines due to over-exploitation, habitat loss, and predation by invasive species were not reflected in the genetic analyses in this study. Few bottleneck effects were detected, even in the Tokashikijima population (O9), which showed the lowest basic genetic values in all populations used in this study (Table 1). As mentioned in the above section, Tokashikijima and southern Okinawajima populations may have maintained gene flow during at least the late Pleistocene; if this occurred, decline of effective population size in Tokashikijima should have progressed slowly after geographic isolation from Okinawajima without a severe bottleneck. A similar scenario could also have occurred in the Ukeshima and Sesokojima populations because these showed ancient gene flow from adjacent populations in Amamioshima and Okinawajima. For conservation, the Tokashikijima population should be the highest priority target. Because of the lower genetic diversity and no present gene flow from other populations, the Tokashikijima population should be more sensitive to reduction in habitat and number of individuals than other populations because of negative genetic influences, such as deleterious effects of inbreeding and loss of genetic diversity (Frankham et al. 2002). Consequently, each genetic population unit that has a distinct ancestral group with scarce gene flow should be recognized as unique. Because environmental changes can often impede natural migration and potential gene flow among these populations, maintenance of a natural environment in this species’ distribution is important for sustainable in situ conservation of E. andersoni. Our data can be a fundamental genetic information for ex situ conservation (e.g., Igawa et al. 2013b), reintroduction and genetic reinforcement for the small threatened populations if it is needed in the future.
References
Bowcock AM, Ruiz-Linares A, Tomfohrde J et al (1994) High resolution of evolutionary trees with polymorphic microsatellites. Nature 368:455–457. https://doi.org/10.1038/368455a0
Campana MG, Hunt HV, Jones H, White J (2011) CorrSieve: software for summarizing and evaluating structure output. Mol Ecol Resour 11:349–352. https://doi.org/10.1111/j.1755-0998.2010.02917.x
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Eldridge MDB, King JM, Loupis AK et al (1999) Unprecedented low levels of genetic variation and inbreeding depression in an island population of the black-footed rock-wallaby. Conserv Biol 13:531–541. https://doi.org/10.1046/j.1523-1739.1999.98115.x
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Felsenstein J (1989) PHYLIP—phylogeny inference package (Version 3.2). Cladistics 5:164–166
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508. https://doi.org/10.1046/j.1523-1739.1996.10061500.x
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity (Edinb) 78(Pt 3):311–327. https://doi.org/10.1038/sj.hdy.6880980
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Garza JC, Williamson EG (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10:305–318
Gilpin M, Soulé M (1986) Minimum viable populations: processes of species extinction. In: Soulé M (ed) Conservation biology: the science of scarcity and diversity. Sinauer Associates, Sunderland, pp 19–34
Giordano AR, Ridenhour BJ, Storfer A (2007) The influence of altitude and topography on genetic structure in the long-toed salamander (Ambystoma macrodactulym). Mol Ecol 16:1625–1637. https://doi.org/10.1111/j.1365-294X.2006.03223.x
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). http://www2.unil.ch/popgen/softwares/fstat.htm
Guillot G, Estoup A, Mortier F, Cosson JF (2005) A spatial statistical model for landscape genetics. Genetics 170:1261–1280. https://doi.org/10.1534/genetics.104.033803
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48:361–372
Hayashi T, Matsui M, Utsunomiya T et al (1992) Allozyme variation in the newt Tylototriton andersoni from three islands of the Ryukyu archipelago. Herpetologica 48:178–184
Hikida T, Ota H (1997) Biogeography of reptiles in the subtropical East Asian Islands. In: Lue KY, Chen TH (eds) Proceedings of the symposium on the phylogeny, biogeography and conservation of fauna and flora of East Asian Region. National Science Council, R. O. C., Taipei, pp 11–28
Hinten G, Harriss F, Rossetto M, Braverstock PR (2003) Genetic variation and island biogreography: microsatellite and mitochondrial DNA variation in island populations of the Australian bush rat, Rattus fuscipes greyii. Conserv Genet 4:759–778. https://doi.org/10.1023/B:COGE.0000006113.58749.ac
Honda M, Matsui M, Tominaga A et al (2012) Phylogeny and biogeography of the Anderson’s crocodile newt, Echinotriton andersoni (Amphibia: Caudata), as revealed by mitochondrial DNA sequences. Mol Phylogenet Evol 65:642–653. https://doi.org/10.1016/j.ympev.2012.07.019
Hou M, Wu Y, Yang K et al (2014) A missing geographic link in the distribution of the genus Echinotriton (Caudata: Salamandridae) with description of a new species from southern China. Zootaxa 3895:89. https://doi.org/10.11646/zootaxa.3895.1.5
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Resour 9:1322–1332. https://doi.org/10.1111/j.1755-0998.2009.02591.x
Igawa T, Okuda M, Oumi S et al (2011) Isolation and characterization of twelve microsatellite loci of endangered Ishikawa’s frog (Odorrana ishikawae). Conserv Genet Resour 3:421–424. https://doi.org/10.1007/s12686-010-9370-7
Igawa T, Oumi S, Katsuren S, Sumida M (2013a) Population structure and landscape genetics of two endangered frog species of genus Odorrana: different scenarios on two islands. Heredity (Edinb) 110:46–56. https://doi.org/10.1038/hdy.2012.59
Igawa T, Sugawara H, Tado M et al (2013b) An attempt at captive breeding of the endangered newt Echinotriton andersoni, from the Central Ryukyus in Japan. Animals 3:680–692. https://doi.org/10.3390/ani3030680
Ikehara S (1996) Islands of valuable animals: fauna of the Ryukyu archipelago. In: Nakamura K, Ujiie H, Ikehara S et al (eds) Nature in Japan, 8. Southern Islands, Iwanami-Shoten, pp 149–160
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806. https://doi.org/10.1093/bioinformatics/btm233
Kan H (2014) Formation process of coral reefs in Ryukyu Islands. In: Takamiya H, Shinzato T (eds) Empirical studies for environmental and cultural change in pre- and proto-historic era, vol 2. Rokuichi Shobo. Tokyo, Japan, pp 19–28
Kaneko Y, Matsui M (2004) Echinotriton andersoni. In: IUCN Red List Threat Species Version 2014 2. http://www.iucnredlist.org/. Accessed 14 Nov 2014
Kizaki K, Oshiro I (1977) Paleogeography of the Ryukyu Islands. Mar Sci Mon 9:542–549
Kizaki K, Oshiro I (1980) The origin of the Ryukyu Islands. In: Kizaki K (ed) Natural history of the Ryukyus. Tsukijishokan, Tokyo, pp 8–37
Lecis R, Norris K (2004) Population genetic diversity of the endemic Sardinian newt Euproctus platycephalus: implications for conservation. Biol Conserv 119:263–270. https://doi.org/10.1016/j.biocon.2003.11.011
Manni F, Guérard E, Heyer E (2004) Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Hum Biol 76:173–190
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Nakamura Y, Ota H (2015) Late Pleistocene-Holocene amphibians from Okinawajima Island in the Ryukyu Archipelago. Reconfirmed faunal endemicity and the Holocene range collapse of forest-dwelling species. Palaeontol Electr 18:1–26. https://palaeo-electronica.org/content/in-press/935-okinawan-amphibians
Nei M, Maruyama T, Chakraborty R (1975) The bottleneck effect and genetic variability in populations. Evolution 29:1–10
Nei M, Tajima F, Tateno Y (1983) Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol 19:153–170. https://doi.org/10.1007/BF02300753
Okamoto T (2017) Historical biogeography of the terrestrial reptiles of Japan: a comparative analysis of geographic ranges and molecular phylogenies. In: Motokawa M, Kajihara H (eds) Species diversity of animals in Japan. Springer, Tokyo, pp 135–163
Osozawa S, Shinjo R, Armid A et al (2012) Palaeogeographic reconstruction of the 1.55 Ma synchronous isolation of the Ryukyu Islands, Japan, and Taiwan and inflow of the Kuroshio warm current. Int Geol Rev 54:1369–1388. https://doi.org/10.1080/00206814.2011.639954
Osozawa S, Su Z-H, Oba Y et al (2013) Vicariant speciation due to 1.55 Ma isolation of the Ryukyu islands, Japan, based on geological and GenBank data. Entomol Sci 16:267–277. https://doi.org/10.1111/ens.12037
Ota H (1998) Geographic patterns of endemism and speciation in amphibians and reptiles of the Ryukyu Archipelago, Japan, with special reference to their palaeogeographical implications. Res Popul Ecol (Kyoto) 40:189–204
Ota H (2000) The curent geographic faunal pattern of reptiles and amphibians of the Ryukyu Archipelago and adjacent regions. Tropics 10:51–62. https://doi.org/10.3759/tropics.10.51
Peakall R, Smouse PE (2006) genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295. https://doi.org/10.1111/j.1471-8286.2005.01155.x
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90:502–503. https://doi.org/10.1093/jhered/90.4.489
R Core Team (2011) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219
Rousset F (2008) genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Saitou N, Nei M (1987) The Neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Savage WK, Fremier AK, Shaffer HB (2010) Landscape genetics of alpine Sierra Nevada salamanders reveal extreme population subdivision in space and time. Mol Ecol 19:3301–3314. https://doi.org/10.1111/j.1365-294X.2010.04718.x
Sugawara H, Igawa T, Yokogawa M et al (2012) Isolation and characterization of ten microsatellite loci of endangered Anderson’s crocodile newt, Echinotriton andersoni. Conserv Genet Resour 4:595–598. https://doi.org/10.1007/s12686-012-9600-2
Takezaki N, Nei M, Tamura K (2010) POPTREE2: Software for constructing population trees from allele frequency data and computing other population statistics with Windows interface. Mol Biol Evol 27:747–752. https://doi.org/10.1093/molbev/msp312
Tamura K, Peterson D, Peterson N et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Tominaga A, Ota H, Matsui M (2010) Phylogeny and phylogeography of the sword-tailed newt, Cynops ensicauda (Amphibia: Caudata), as revealed by nucleotide sequences of mitochondrial DNA. Mol Phylogenet Evol 54:910–921. https://doi.org/10.1016/j.ympev.2009.11.002
Utsunomiya Y, Utsunomiya T, Kawachi S (1978) Some ecological observations of Tylototriton andersoni, a terrestrial salamander occurring in the Tokunoshima Island. Proc Japan Acad Ser B Phys Biol Sci 54:341–346. https://doi.org/10.2183/pjab.54.341
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Wang IJ, Savage WK, Shaffer HB (2009) Landscape genetics and least-cost path analysis reveal unexpected dispersal routes in the California tiger salamander (Ambystoma californiense). Mol Ecol 18:1365–1374. https://doi.org/10.1111/j.1365-294X.2009.04122.x
Wang S, Zhu W, Gao X et al (2014) Population size and time since island isolation determine genetic diversity loss in insular frog populations. Mol Ecol 23:637–648. https://doi.org/10.1111/mec.12634
White TA, Searle JB (2007) Genetic diversity and population size: island populations of the common shrew, Sorex araneus. Mol Ecol 16:2005–2016. https://doi.org/10.1111/j.1365-294X.2007.03296.x
Acknowledgements
We thank Noriko Iwai and Kazuki Yamamuro for their field assistance. Echinotriton andersoni was designated as a natural monument by Kagoshima and Okinawa Prefectural Governments, and its handling is thus regulated by law. Our sampling was conducted with permission from the Boards of Education of both governments. Moreover, sampling was completed prior to designation of this newt as a rare species of wild fauna and flora of Japan by the national government in 2016.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (B and C) (Grant Nos. 24310173, 20510216) to M. S. and a Grant-in-Aid for Young Scientists (B) (Grant Nos. 23710282, 26830144) to T. I. from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Igawa, T., Sugawara, H., Honda, M. et al. Detecting inter- and intra-island genetic diversity: population structure of the endangered crocodile newt, Echinotriton andersoni, in the Ryukyus. Conserv Genet 21, 13–26 (2020). https://doi.org/10.1007/s10592-019-01219-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01219-8