
Abstract
Kichulchoia multifasciata, a small-sized loach (Family Cobitidae) inhabiting only the Nakdong River on the Korean Peninsula, is generally found around well-oxygenated minor tributaries. Despite the scarcity, this species is not yet a legally protected species in South Korea, and the spatial pattern of genetic diversity, which is essential to create management strategies, has never been studied. In this study, three mitochondrial loci and ten microsatellites were used to analyze the genetic diversity and the structure among eight K. multifasciata populations collected from different tributaries. K. multifasciata populations showed a high level of mitochondrial variability, with above 0.9 of average haplotype diversity in all three loci analyzed. A substantial microsatellite polymorphism was also found. Weak genetic structuring among populations and isolation by distance were clearly revealed in both mitochondrial and microsatellite data, indicating the high levels of gene flow among the tributaries. However, a few of the populations exhibited a genetic signature that they have experienced the historical size decline. Our data suggest that unequal gene flow might be one of the causes; nevertheless, the possibility of effective population size reduction by other factors could not be completely excluded. Our genetic evidence can be the critical information needed to protect the populations from the genetic erosion in the Nakdong River that has been destroyed and fragmented with increasing anthropogenic interferences.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The freshwater fish provides excellent model systems for studying historical processes of geographic isolation and population genetic differentiation, since the habitats are generally composed of physically disconnected water bodies. Because of this insular nature, genetic differentiation among populations in freshwater fish species is generally greater than that observed in seawater or terrestrial species (reviewed by Ward et al. 1994; DeWoody and Avise 2000). Even within a single water body, gene flow among populations may also be restricted due to at least two factors. The first factor is natural landscape structures, like waterfalls and valleys (e.g. Castric et al. 2001; Crispo et al. 2006; Caldera and Bolnick 2008).
The second factor is adaptive divergence under variable selection pressures. In a species that have a favorable trait in a particular environment, residents tend to have higher fitness than dispersers do (e.g. Hecht et al. 2015; Brauer et al. 2016; Hand et al. 2016; reviewed by; Schluter 2000; Hendry 2004). Assuming this, for example, it may be uncommon for species inhabiting an upstream region to migrate through the main stem into other upstream regions. In such species, thus, the level of historical gene flow between populations in different tributaries are expected to be correlated with the length of the main stem connecting the tributaries, and genetic differentiation among populations is likely to occur in the pattern of isolation by distance (e.g. Griffiths et al. 2009; Crookes and Shaw 2016). It should also be noted that gene flow may occur due to fusion between headwaters (e.g., Schönhuth et al. 2011, 2012). In such case, genetic differentiation between populations becomes correlated with the straight distance between headwaters or populations.
The Korean Peninsula is a small area located in the south tip of East Asia, but it contains an extremely high endemic freshwater species (67 species only in South Korea; approximately 35%) probably due to its geographical complexity reflecting the past geological history (Yoon et al. 2018). Kichulchoia multifasciata is also one of the representative endemic species on the Korean Peninsula (Kim and Lee 1995; Kim and Yang 2016). K. multifasciata, the member of 15 South Korean cobitids, is endemic to the Nakdong River, a 510 km long drainage system running through the south eastern corner of South Korea (Kim and Lee 1995; Kim and Yang 2016). This species is generally found around well-oxygenated upstream areas in minor tributaries, characterized by pool-riffle sequences with substrates being composed of small rocks and gravels (Kim and Yang 2016). The physical feature observed in the main stem of this river is drastically different from that of the upstream areas. For example, the main stem is substantially wider, normally consists of fine sandy bottoms, and has extremely slower water flow. Assuming that this species is well adapted to the environmental features of upstream areas in tributaries, the migration among populations in different tributaries is expected to be scarce, even though there is no big natural landscape structure that restricts the movement of individuals in the main stem of this river. However, the genetic diversity and structure of this species has not yet been studied.
In this study, we used three mitochondrial and ten microsatellite loci to determine the intrapopulation genetic diversity and the level of spatial genetic structure of eight K. multifasciata populations collected from different tributaries in the Nakdong River. In particular, we attempted to test whether the spatial distance affected the historical level of gene flow between K. multifasciata populations. In addition, we attempted to investigate whether populations showed signatures that had undergone historical or contemporary changes in size. Given that this species is native to this river all over the world, the genetic diversity of this species is expected to be very low, and systematic management plans should be established for the conservation. The genetic results of this study are anticipated to provide an important insight into the establishment of conservation strategies for this species as well as other endemic freshwater species restricted to a single drainage.
Materials and methods
Sampling and DNA extraction
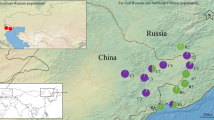
Sampling of Kichulchoia multifasciata was performed at eight spots (populations) throughout the Nakdong River (Fig. 1) according to the Inland Water Fisheries Act and Wildlife Protection and Management Act of the Republic of Korea. Each spot was located in different tributaries from the Nakdong drainage. A total of 256 adults were caught between September 2013 and July 2015 using a kick-net (0.5 mm × 0.5 mm mesh). The whole procedure of collection, tissue sampling and experimental process was approved by the Yeungnam University Institutional Animal Care and Use Committee (Protocol # 2015013). The tip of caudal fin (1 × 1 mm) was briefly cut from each individual and stored in 95% ethanol, and all individuals collected were released back to the original collection sites. Genomic DNA was isolated from the fin tissues using Wizard Genomic DNA Purification Kit (Promega, Madison, WI) according to manufacturer’s instructions.

a The sampling sites of Kichulchoia multifasciata populations analyzed in this study. The Nakdong River was highlighted by blue colored line and each sampling site (population) was represented with solid circle; YR, 128°00′29.9″E, 36°35′45.2″N; GA, 128°53′51.0″E, 36°26′37.3″N; WR, 128°46′44.0″E, 36°07′44.0″N; GH, 128°57′50.0″E, 35°57′56.5″N; NR, 128°43′32.2″E, 35°47′26.5″N; HR, 127°54′43.6″E, 35°41′04.2″N; CD, 128°45′53.6″E, 35°35′21.5″N; YA, 127°59′26.2″E, 35°17′26.9″N. The location of the Korean Peninsula was indicated in the map placed on the left above. b The photo of K. multifasciata. (Color figure online)
Mitochondrial sequencing
Three mitochondrial loci, cytochrome oxidase subunit I (COI), cytochrome b (cyt b) and control region (CR) were analyzed for all collected K. multifasciata individuals. Three previously reported primer sets were used for the amplification of each locus (COI: Folmer et al. 1994; CR:; Liu et al. 2002; cyt b:; Xiao et al. 2001; Table S1 in Supplementary material). For the PCR amplification, Piko Thermal Cycler (Finnzymes, Espoo, Finland) was used with a 25 µL reaction mixture containing 1 µL genomic DNA (10–50 ng), 1X Taq buffer, 5 pmole of dNTP, 5 pmole of each primer and 2.5 unit of Prime Taq DNA polymerase (GenetBio, Daejeon, South Korea). Thermal cycling was composed of denaturation at 94 °C for 10 min followed by 35 cycles of 94 °C for 30 s, 54–58 °C for 30 s and 72 °C for 30 s and final extension at 72 °C for 10 min. The PCR products were purified using Primeprep PCR Purification Kit (GenetBio, Daejeon, South Korea) and were commercially sequenced using the PCR primers on an ABI 3730XL DNA Analyzer with BigDye terminator system (Applied Biosystems, Foster City, CA) at Biomedic Inc (Bucheon, South Korea).
Microsatellite genotyping
All collected fin-clip samples were also used in microsatellite genotyping with ten previously published primer sets (CL: Kwan et al. 2014; Cota: De Gelas et al. 2008; IC: Bang et al. 2009; Table S1 in Supplementary material). Each forward primer was fluorescently labeled with FAM, HEX, NED or PET (Applied Biosystems). Amplification protocols were the same as described above (Mitochondrial sequencing). The fluorescently labeled PCR products were genotyped at Biomedic Inc using an ABI 3730 XL Genetic Analyzer. Alleles were scored using GeneMapper 3.7 (Applied Biosystems).
Mitochondrial analyses
All mitochondrial sequences obtained were confirmed through BLAST searches and aligned using MEGA 6.06 (Tamura et al. 2013). The coding loci, cyt b and COI were rechecked against the inferred reading frame for the corresponding protein. All haplotypes obtained in this study were deposited in NCBI GenBank with Accession NOs KY423874 to KY424053. The intrapopulation genetic diversity was quantified based on the number of haplotypes (h), haplotype diversity (hd; Nei 1987), and nucleotide diversity (π; Nei 1987) estimated using DnaSP 5.10 (Librado and Rozas 2009).
Three different approaches were applied to obtain the best approximation regarding the demographic history of K. multifasciata populations. First, two neutrality indices, Tajima’s D (Tajima 1989) and Fu’s Fs (Fu 1997) were calculated using DnaSP to examine whether our haplotype data deviated from the expected values obtained under the neutral model and the assumption of demographic equilibrium. A significant negative value in each index signifies a demographic expansion, whereas a significant positive value signifies a recent demographic decline or a strong population structure. Second, the observed distribution of pairwise differences between sequences was visualized for the major clades obtained (mismatch distribution analysis) under a model of population expansion (Rogers and Harpending 1992) using Arlequin 3.5 (Excoffier and Lischer 2010). Unimodal distribution is usually shown for populations that have recently experienced a demographic expansion, whereas ragged or multimodal distributions are typically shown for populations at stationary demographic equilibrium for a long time. Finally, Bayesian skyline plot was reconstructed using BEAST 1.8.0 for every population (Drummond et al. 2012) to evaluate the shape of demographic growth over time.
The level of genetic divergence between populations was estimated by the calculation of genetic differentiation index (ФST) between each pair of populations using Arlequin. The significance of each ФST value was tested using 1000 permutations of the data. GenAlEx 6.5 (Peakall and Smouse 2012) was used to statistically evaluate the relationship between pairwise-ФST and the interpopulation geographic distance along the length of the main stem. Haplotype network was constructed for each mitochondrial locus and for the combined data, based on median-joining algorithm using PopART 1.7 (Leigh and Bryant 2015), to analyze the distributional pattern of haplotype diversity.
Microsatellite analyses
The level of microsatellite genetic diversity was evaluated by computing the mean (for each population) or total (for each locus) number of alleles per locus (A), allelic richness (AR), observed (HO) and expected (HE) heterozygosity and fixation index (FIS) for each population and locus using Arlequin and Fstat 2.9.3.2 (Goudet 2001). The deviation of observed genotype proportion from that expected under Hardy–Weinberg equilibrium (HWE) was tested for each locus-population pair based on the Fisher’s exact test following Markov chain parameters with 1000 batches and 10,000 iterations per batch (Guo and Thompson 1992), which was implemented in the Genepop 4.2 (Raymond and Rousset 1995). The relative proportion of family relationship (full-sib, half-sib and parental-offspring) was calculated for all pairs of individuals within each population to check the level of inbreeding using ML-Relate (Kalinowski et al. 2006) and COLONY 2.0 (Jones and Wang 2010).
The signature of recent bottleneck was examined by testing the significant excess of expected heterozygosities under the mutation-drift equilibrium relative to those under HWE in Bottleneck 1.2.1 (Piry et al. 1999). The significance was determined based on the Wilcoxon signed-rank test under two-phase model (TPM; 70% stepwise mutation model (SMM) + 30% infinite allele model (IAM)). The signature of population bottleneck was also checked using the same software based on a mode-shift away from the typical L-shaped distribution of allelic frequencies (Luikart et al. 1998). The signature of historical bottleneck was tested by the calculation of M-ratio, the mean ratio of the number of alleles to the size range of alleles (Garza and Williamson 2001) in Arlequin. Critical_M (Garza and Williamson 2001) was used to estimate the threshold value of M ratio (MC) for each population; the population that has experienced historical decline in their size are expected to display lower M ratio than the MC. For each population MC was estimated with two different values of θ (= 4 Neµ), 1 and 10, representing 500 and 5000 of population size before decline, respectively, and default options for other values (Δg, the mean size of non-one-step changes = 3.5; PS = 0.9).
To estimate the level of genetic structure among K. multifasciata populations, we attempted to use two different methods. First, the calculation of pairwise-FST and -RST values was implemented in Arlequin. The significances of pairwise-FST and -RST values were determined through 1000 permutations. GenAlEx was used to statistically evaluate the relationship between pairwise-FST and -RST values and the interpopulation geographic distance along the length of the main stem. Not only the length of the stream, but also the linear distance between the populations and the headwater distance were also measured. The distance was estimated using Googlemap’s ‘Measure Distance’ function. Second, the existence of genetic cluster was predicted based on Bayesian framework on Structure 2.3.4 (Pritchard et al. 2000). This analysis was performed under the admixture model for cluster (K) from one to eight, with 20 independent runs, each consisting of 4 × 105 MCMC generations after burn-in of 105 MCMC generations. The most reliable number of genetic clusters was estimated using the delta K method described by Evanno et al. (2005), which was implemented in Structure Harvester 0.6.94 (Earl and von Holdt 2012).
Results
Kichulchoia multifasciata populations showed a high level of mitochondrial diversity, with above 0.9 in average haplotype diversity of all three loci analyzed (Table 1); a total of 180 mitochondrial haplotypes (COI: 40; cyt b: 60; CR: 80) were found from 256 individuals. Populations HR, WR and YA displayed a slightly lower level of haplotype diversity than other populations, with ranging from 0.723 to 0.738, from 0.797 to 0.843 and from 0.695 to 0.812 in COI, cyt b and CR, respectively (Table 1). The nucleotide diversity was also lower in these three populations than in other populations (Table 1). The sequences of COI and cyt b showed significant deviations from the neutral expectation estimated based on both Tajima’s D and Fu’s Fs, while CR showed the significance only in Fu’s Fs (Table 1), indicating the signature of drastic demographic expansion following the colonization of this species. At the population level, the signature of drastic demographic expansion was only found in populations GA, GH and YR (Table 1). Our Bayesian skyline plots showed that the effective population sizes of K. multifasciata greatly increased around 0.2–0.6 My (Fig. 2). A noticeable increase was found in all populations except for populations HR, WR and YA (Fig. 2).

Extended Bayesian skyline plots (EBSPs) implemented in BEAST to evaluate the shape of each population growth over historical time
A substantial microsatellite polymorphism was also observed from the 10 loci examined with the number of alleles per loci ranging from 10 (IC654) to 43 (CL002) and the observed and expected heterozygosities ranging from 0.561 (CL372) to 0.920 (CL002) and from 0.569 (CL372) to 0.934 (IC248), respectively (Table 2). CL010 was the only locus showing the significant deviation (p = 0.002) from HWE expectation following Bonferroni adjustment (n = 10; α = 0.005; Table 2). However, this locus was included in the subsequent analyses, because the deviation seemed to be caused from the deficit of observed heterozygosity only in populations NR, HR and YA. Similarly to the results from mitochondrial analyses, the allelic diversity of microsatellites was lowest in populations HR, WR, and YA (Table 3). Our maximum likelihood analyses of relatedness using ML-Relate and Colony showed consistently low levels of family relationship in all populations (ML-Relate: 7.9–17.6%; COLONY: 6.3–8.7%; Table 3). No evidence of contemporary population bottleneck could be found from the test with mode-shift in allele class distribution (Table 3). However, population WR showed a significance in Wilcoxon signed-rank test in Bottleneck following Bonferroni adjustment (n = 8, α = 0.00625; Table 3). M-ratio values of K. multifasciata populations varied between 0.650 and 0.823. Populations HR and WR showed the M-ratio values significantly less than Mc (Fig. 3), indicating the signature of historical demographic decline in these populations.

M ratio values calculated from microsatellite genotyping. Each circle indicates the average M ratio of each Kichulchoia multifasciata population with standard deviation. The dashed line indicates the Mc threshold (max and min) calculated from parameter ɵ = 1 to ɵ = 10
Haplotypes of K. multifasciata (from the combined data of three mitochondrial loci) could likely be allocated into four different clades in our median-joining network analysis, though the level of divergence was not large between any pair of clades (Fig. 4). No geographic pattern was observed, because each clade was represented by most of the populations examined (Fig. 4). In the mismatch analyses, clade 1 and 4 exhibited clear unimodal distribution, whereas multimodal distribution was observed in clade 2 and 3 (Fig. 4). In pairwise comparison of ФST, no population structure was found (Table 4). However, population HR displayed relatively greater genetic differentiation from others (0.093–0.201; Table 4). Pairwise values of ФST were positively correlated with the (main stem) geographic distances between populations (Fig. 5a).

Median-joining network reconstructed based on the haplotypes of three mitochondrial loci (COI, cyt b and CR) obtained from the eight Kichulchoia multifasciata populations. The results of mismatch analyses showing the past population demographics were added for the four clades identified in this network

Plots of the isolation by distance (IBD) analysis of the eight population of Kichulchoia multifasciata. a Regression between mitochondrial genetic differentiation index (pairwise-ФST) and interpopulation geographic distance (km). b Regression between microsatellite pairwise-FST or -RST values and interpopulation geographic distances (km)
The pairwise comparisons of FST and RST were very low and many of them were not statistically significant (Table 4). Similarly to our mitochondrial results, isolation by distance was also observed in our pairwise-FST and -RST estimates (Fig. 5b). However, straight distances between headwaters or populations did not show any significant correlation with pairwise ФST values (headwater: R2 = 0.0026; populations: R2 = 0.0006) or pairwise comparisons of FST (headwater: R2 = 0.0070; populations: R2 = 0.0051) and RST values (headwater: R2 = 0.0002; populations: R2 = 0.0048). Our Bayesian clustering analysis using Structure revealed the absence of a strong genetic structuring among K. multifasciata populations (Fig. 6). The delta K method implemented in Structure Harvester indicated that the optimal number of clusters might be two or six (Fig. 6a). At K = 2, the assignment histogram was displayed as a completely admixed pattern, whereas populations HR and WR were slightly separated from others at K = 6 (Fig. 6b).

Population structure of Kichulchoia multifasciata estimated from microsatellite genotyping. a The estimation of most reliable number (2 or 6) of cluster based on the delta K method. b The population structure patterns obtained based on K = 2 (above) or 6 (below). Each cluster was denoted by different colors. (Color figure online)
Discussion
The basic aim of this study was to estimate the level of genetic diversity and historical gene flow among populations in Kichulchoia multifasciata. A considerable genetic polymorphism was detected at microsatellite loci examined. According to a meta-study performed by DeWoody and Avise (2000), the mean number of alleles per locus and the mean heteozygosity in microsatellites of freshwater fish species are 0.46 and 7.5, respectively. However, the overall genetic diversity estimates of K. multifasciata populations were substantially higher than these values, with 0.82 of observed heterozygosity and 11.9 of mean allele number per locus. Such a high degree of polymorphism was also present in the mitochondrial loci examined in this study. Total number of haplotypes and mean haplotype diversity were 114 and 0.99, respectively, when all three loci were combined. It is uncommon to expect for a species living only in a river that is not very large in the world to exhibit such a high genetic diversity.
The fact that this species with a limited distribution has such a considerable level of polymorphism suggests that new alleles have been generated at a fairly rapid rate since its colonization, and/or that the natural conditions allowing the populations to maintain their size well were established in this river. The results of the haplotype network analysis indicated the process of rapid genetic differentiation and population expansion that occurred after the early establishment of this species to the Nakdong River. Expansion and genetic differentiation were observed to occur intensively in the haplotype clades 1 and 4, suggesting that these clades were probably more common alleles rather than 2 and 3 in the early stage. This pattern was strongly supported by our results of mitochondrial demographic statistics (mismatch distribution analysis). Since each clade contains the haplotypes of almost all populations, these four clades may have existed from the time of colonization or differentiated at the very beginning of the expansion. Although we can not exclude the hypothesis that these clades were rather later created by spatial isolation and then mixed by active gene flows, no signature of spatial variation was observed in our clade distribution data.
Considering the conditions of the region where K. multifasciata is presently found, the most plausible reason for the rapid expansion of the early colonized population is that there was no competitor of this species occupying similar ecological niche and that there were not many predators directly targeting this species. However, since the behavior of this species has not yet been examined in the natural ecosystem, such a speculation requires further cautions. Although not as a direct concern of this study, investigation regarding the phylogenetic placement of this species among its congeneric or cofamilial taxa and the biogeodispersal process of this species may provide insight into the initial colonization process. Since congeneric species and the closely related genus (e.g. Niwaella) are sporadically distributed throughout East Asia (Kim and Lee 1995; Chen and Chen 2005; Kim and Yang 2016), a fairly wide range of work will be required to examine the phylogeographic processes. However, according to a recent phylogenetic study, Kichulchoia seemed to be more closely associated with Iksookimia than Niwaella (Perdices et al. 2016), and there is also a need to pay attention to the relationship with other cofamilial species inhabiting the Korean Peninsula.
It is conceivable that K. multifasciata populations might not have experienced any significant reduction or allelic loss, simply given that all populations examined had substantially high genetic diversity. Our analytical results also provided evidence supporting this proposition. First, our extended Bayesian skyline plots showed that the effective size of all K. multifasciata populations has increased, though the degree of increase was different among populations. Second, all populations showed quite low frequency of family relationships among individuals. Finally, no population showed a complete signature of drastic decline in our genetic bottleneck analysis. Based on the weak spatial genetic structuring and isolation by distance among populations in our results, active gene flows among populations may have played a role, at least in part, in maintaining population size.
As mentioned in Introduction, however, active gene flows were not much expected among populations located in different tributaries past deep and sandy main stem of the river. Even if this species is strongly adapted to the upstream environment, our results claimed that it may not be right to assume that migrating through the main stem is not feasible. K. multifasciata spends most of the time dragging on the floor and does not swim often. This locomotion habit may serve as a good strategy to avoid predation in an open environment of main stem area. This suggests that the Nakdong River, which has little natural landscape structure that interrupts the migration, provides a suitable condition for this species to maintain gene flow among the populations and expand the habitat area.
Populations HR, WR and YA exhibited relatively lower levels of genetic diversity than others in the mitochondrial loci analyzed. A similar distributional pattern of genetic diversity appeared in our microsatellite data. Specifically, populations HR and WR showed a signature of historical size decline, since they had average M ratios lower than the critical values calculated for the individual populations. These two populations were also distinctly prominent in the Structure analysis. The mean mitochondrial ФST values of the populations HR, WR and YA with other populations were remarkably greater than those of other populations (Table 4), suggesting that the populations were relatively isolated in the gene flow network. This comparison was not possible in microsatellites, because most of the pairwise FST and -RST values were not statistically significant. It is difficult to conclude that the gene flow reduction of these populations is necessarily due to distances from other populations. None of the populations were distantly isolated. Therefore, we suggest that ecological studies are needed to see if the waterways into these populations differ from other areas in the river. Other factors besides gene flow can also be considered to explain the unequal distribution of genetic diversity. The size of the effective populations has been possibly reduced, due to the lack of environmental conditions to sustain large populations, and that the resulted genetic drift has reduced the genetic similarities with other populations.
Only considering the distribution of K. multifasciata in the world, it is absolutely not too much to designate this species as a species under special protection. Probably, this species seems to have not been included in the protected species list of the South Korean government, only because it is a species commonly observed in the Nakdong River. However, as our results showed, the population size and genetic diversity of this species could have been maintained due to the various factors conditioned by the natural environment of the river. However, this river has been completely cut off due to artificial structures constructed over the last decades and gene flow among different tributaries is not possibly achieved any more. Under this circumstance, some populations, which were noted in our study, are likely to suffer from the genetic erosion even in a short period of time (Reed and Frankham 2003; Laroche and Durand 2004). Our genetic data can provide the critical information on the timing when individual translocation is required or when the naturalization of the river system needs to be performed.
References
Bang IC, Kim WJ, Lee IR (2009) Characterization of polymorphic microsatellite loci in the endangered Miho spine loach (Iksookimia choii) and cross-species amplification within the Cobitidae family. Mol Ecol Resour 9(1):281–284
Brauer CJ, Hammer MP, Beheregaray LB (2016) Riverscape genomics of a threatened fish across a hydroclimatically heterogeneous river basin. Mol Ecol 25(20):5093–5113
Caldera EJ, Bolnick DI (2008) Effects of colonization history and landscape structure on genetic variation within and among threespine stickleback (Gasterosteus aculeatus) populations in a single watershed. Evol Ecol Res 10:575–598
Castric V, Bonney F, Bernatchez L (2001) Landscape structure and hierarchical genetic diversity in the brook charr, Salvelinus fontinalis. Evolution 55(5):1016–1028
Chen Y, Chen Y (2005) Revision of the genus Niwaella in China (Pisces, Cobitidae), with description of two new species. J Nat Hist 39(19):1641–1651
Crispo E, Bentzen P, Reznick DN, Kinnison MT, Hendry AP (2006) The relative influence of natural selection and geography on gene flow in guppies. Mol Ecol 15(1):49–62
Crookes S, Shaw PW (2016) Isolation by distance and non-identical patterns of gene flow within two river populations of the freshwater fish Rutilus rutilus (L. 1758). Conserv Genet 17(4):861–874
De Gelas K, Janko K, Volckaert FAM, De Charleroy D, Van Houdt JKJ (2008) Development of nine polymorphic microsatellite loci in the spined loach, Cobitis taenia, and cross-species amplification in the related species C. elongatoides, C. taurica and C. tanaitica. Mol Ecol Resour 8(5):1001–1003
DeWoody JA, Avise JC (2000) Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J Fish Biol 56(3):461–473
Drummond AJ, Suchard MA, Xie D, Rambaut A (2012) Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29(8):1969–1973
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14(8):2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3(5):294–299
Fu Y (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147(2):915–925
Garza J, Williamson E (2001) Detection of reduction in population size using data from microsatellite loci. Mol Ecol 10(2):305–318
Goudet J (2001) FSTAT, a program to estimate and test gene diversity and fixation indices (version 2.9.3). http://www2.unil.ch/popgen/softwares/fstat.htm
Griffiths AM, Koizumi I, Bright D, Stevens JR (2009) A case of isolation by distance and short-term temporal stability of population structure in brown trout (Salmo trutta) within the River Dart, southwest England. Evol Appl 2(4):537–554
Guo SW, Thompson EA (1992) Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48(2):361–372
Hand BK, Muhlfeld CC, Wade AA, Kovach RP, Whited DC, Narum SR, Matala AP, Ackerman MW, Garner BA, Kimball JS, Stanford JA, Luikart G (2016) Climate variables explain neutral and adaptive variation within salmonid metapopulations: the importance of replication in landscape genetics. Mol Ecol 25(3):689–705
Hecht BC, Matala AP, Hess JE, Narum SR (2015) Environmental adaptation in Chinook salmon (Oncorhynchus tshawytscha) throughout their North American range. Mol Ecol 24(22):5573–5595
Hendry AP (2004) Selection against migrants contributes to the rapid-evolution of reproductive isolation. Evol Ecol Res 6:1219–1236
Jones OR, Wang J (2010) COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10(3):551–555
Kalinowski ST, Wagner AP, Taper ML (2006) ML-Relate: a computer program for maximum likelihood estimation of relatedness and relationship. Mol Ecol Resour 6(2):576–579
Kim IS, Lee WO (1995) Niwaella brevifasciata, a new cobitid fish (Cypriniformes: Cobitidae) with a revised key to the species of Niwaella. Jap J Ichthyol 42(3/4):285–290
Kim HS, Yang H (2016) Spawning period and spawning characteristics of Kichulchoia multifasciata (Pisces: Cobitidae) in the Yugokcheon (stream) of Nakdonggang (river) from Korea. Kor J Ichthyol 28(2):93–99
Kwan YS, Ko MH, Won YJ (2014) Genomic replacement of native Cobitis lutheri with introduced C. tetralineata through a hybrid swarm following the artificial connection of river systems. Ecol Evol 4(8):1451–1465
Laroche J, Durand JD (2004) Genetic structure of fragmented populations of a threatened endemic percid of the Rhône river: Zingel asper. Heredity 92(4):329–334
Leigh JW, Bryant D (2015) POPART: full-feature software for haplotype network construction. Methods Ecol Evol 6(9):1110–1116
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25(11):1451–1452
Liu H, Tzeng CS, Teng HY (2002) Sequence variations in the mitochondrial DNA control region and their implications for the phylogeny of the Cypriniformes. Can J Zool 81(12):1938–1946
Luikart G, Allendorf F, Cornuet J, Sherwin W (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Hered 89(3):238–247
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28(19):2537–2539
Perdices A, Bohlen J, Šlechtová V, Doadrio I (2016) Molecular evidence for multiple origins of the European spined loaches (Teleostei, Cobitidae). PLoS ONE 11(1):e0144628
Piry S, Luikart G, Cornuet J (1999) BOTTLENECK: a computer program for detecting recent reductions in the effective population size using allele frequency data. J Hered 90(4):502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155(2):945–959
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86(3):248–249
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17(1):230–237
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9(3):552–569
Schluter D (2000) The ecology of adaptive radiation. Oxford University Press, Oxford
Schönhuth S, Blum MJ, Lozano-Vilano L, Neely DA, Varela-Romero A, Espinosa H, Perdices A, Mayden RL (2011) Inter-basin exchange and repeated headwater capture across the Sierra Madre Occidental inferred from the phylogeography of Mexican stonerollers. J Biogeogr 38(7):1406–1421
Schönhuth S, Hillis DA, Neely DA, Lozano-Vilano L, Perdices A, Mayden RL (2012) Phylogeny, diversity, and species delimitation of the North American round-nosed minnow (Teleostei: Dionda) as inferred from mitochondrial and nuclear DNA sequences. Mol Phylogenet Evol 62(1):427–446
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30(12):2725–2729
Ward RD, Woodwark M, Skibinski DOF (1994) A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol 44(2):213–232
Xiao W, Zhang Y, Liu H (2001) Molecular systematics of Xenocyprinae (Teleostei: Cyprinidae): taxonomy, biogeography, and coevolution of a special group restricted in East Asia. Mol Phylogenet Evol 18(2):163–173
Yoon JD, Kim JH, Park SH, Jang MH (2018) The distribution and diversity of freshwater fishes in Korean Peninsula. Kor J Ecol Environ 51(1):71–85
Acknowledgements
Dong-Young Kim, Hari Won, and Seul-Ki Park assisted with field or laboratory work. Research supported by a Grant of the National Institute of Biological Resources (NIBR #201403201) funded by the Ministry of Environment and the grant from the National Research Foundation (2015R1D1A2A01058987) in Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bae, HG., Nightingale, C., Jeoung, D.H. et al. Genetic diversity and population structure of Kichulchoia multifasciata in South Korea. Conserv Genet 20, 477–487 (2019). https://doi.org/10.1007/s10592-019-01147-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01147-7