
Abstract
Examining the spatial genetic structure of cryptic species occupying challenging terrain can afford otherwise unattainable insights into ecological and evolutionary processes, such as population dynamics and dispersal patterns; information important for optimising conservation management. Using 13 microsatellite markers, we evaluated patterns of fine-scale gene flow and the spatial extent of genetic structuring of rock wren (Xenicus gilviventris), a threatened alpine passerine endemic to mountainous regions of the South Island, New Zealand. Through spatial autocorrelation analysis, we found that the ‘genetic patch size’, i.e. the distance over which individuals were not genetically independent, was surprisingly large (c. 70 km), given the rock wren’s limited flight ability. By estimating recent migration rates among sampling locations we also found asymmetries in gene flow indicative of source–sink dynamics. An area with intensive deer and predator control, in the Murchison Mountains, Fiordland, appears to be a particularly important source of migrants for other populations. These findings suggest that management to maintain connectivity is required across relatively large spatial scales and source populations may be those where introduced mammals are managed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding dispersal dynamics and connectivity among populations of threatened species is crucial in determining the appropriate units and spatial scale for conservation management (Waples and Gaggiotti 2006; Funk et al. 2012). Genetic structuring is typically low in birds and expected to occur only at large geographic spatial scales due to a high capacity for dispersal (Crochet 2000; Abbott et al. 2002). In species where flight is limited however, we might expect fine-scale structuring to occur over small spatial scales of just a few kilometres, more closely resembling the spatial structure of small mammals (Baker et al. 1995; Peakall et al. 2003). Furthermore, avian species that occupy naturally fragmented habitats, such as alpine ‘sky islands’ (Bech et al. 2009) have been demonstrated to exhibit strong genetic structuring (Segelbacher and Storch 2002; Caizergues et al. 2003; Weston and Robertson 2015). The spatial heterogeneity of the alpine landscape and patchiness of suitable habitat may also lead to source–sink population dynamics (O’Keefe et al. 2009). In ‘sink’ populations, reproduction is insufficient to balance local mortality; however, these populations may be sustained through immigration from more productive ‘source’ populations (Pulliam 1988). Source–sink dynamics have important implications for conservation management and can be recognised through asymmetries in dispersal and the effects on genetic diversity within and among populations (King et al. 2000; Andreasen et al. 2012; Furrer and Pasinelli 2015).
Genetic structuring may also arise from a sex-bias in dispersal. Several hypotheses have been proposed to account for differences in dispersal behaviour between the sexes: competition for resources (Greenwood 1980), competition for mates (Perrin and Mazalov 2000) and inbreeding avoidance (Pusey 1987). In birds, females tend to disperse further from their place of birth whilst males exhibit stronger philopatry (i.e. remain at their natal site to breed) (Parkes et al. 1978; Greenwood 1980). This is a prediction of the ‘resource competition’ hypothesis, which states that males are likely to benefit more than females from familiarity with their birth site, as they need to acquire and defend territories to attract mates (Greenwood 1980). Consequently, it can be expected that stronger fine-scale genetic structuring will generally be detected for males in birds.
Here, we investigate the spatial structuring of rock wren (Xenicus gilviventris), a threatened alpine passerine belonging to the endemic New Zealand wren family (Acanthisittidae). Rock wren are a widespread, naturally fragmented taxon, occurring in patches of suitable habitat over c. 900 m in altitude throughout the length of the South Island of New Zealand (Higgins et al. 2001). Several areas of high rock wren density have been identified in localised areas of the lower South Island; areas which have consequently become the focus for conservation management efforts. However, the genetic status of these ‘populations’ and the extent to which they are connected by dispersal has until now, been entirely unknown. By gaining an understanding of the spatial genetic structuring of rock wren and their patterns of fine-scale gene flow, we aim to identify appropriate populations for conservation management. Specifically, we will compare levels of genetic diversity and differentiation among sampling locations, identify any potential asymmetries in gene flow and discuss the implications of these factors for management.
Recent phylogeographic analysis (Weston and Robertson 2015) identified two genetically distinct lineages of rock wren, each spanning approximately 370 km, within which only relatively shallow sub-structuring was detected. This finding, combined with a strong pattern of isolation by distance (Weston and Robertson 2015) suggests that dispersal between geographically isolated areas is occurring, in spite of the rock wren’s poor flight ability. Rock wren are socially monogamous, maintaining territories ranging in size from c. 0.6 to 4.2 hectares (around 1.4 hectares on average), which vary little in size between seasons (Michelsen-Heath 1989). Typical habitat comprises low alpine and subalpine shrubland, herb fields, scree slopes, boulder fields and rocky bluffs (Michelsen 1982). Juveniles establish territories and find mates by the end of the summer in which they fledge, ready to commence breeding the following spring (Michelsen-Heath 1989). Michelsen-Heath (1989) reported that of 75 juveniles banded as nestlings within the Murchison Mountains, Fiordland, 75 % remained within 500 m of their place of fledging, with the remainder not re-sighted (sex bias and time interval were not recorded). Due to their cryptic nature and challenging alpine habitat, very little is known with regard to the dispersal pathways of rock wren, though they have almost never been recorded below the treeline (Weston and Robertson 2015).
Using 13 polymorphic microsatellite DNA loci, we apply both individual and population-level analyses to investigate fine-scale patterns of dispersal in rock wren. We aim to: (i) establish the spatial scale of non-random genetic structure, i.e. the ‘genetic patch size’ of rock wren; (ii) investigate source–sink dynamics, and; (iii) identify the most appropriate populations for conservation management within the lower South Island of New Zealand.
Materials and Methods
Sampling and DNA extraction
Rock wren blood samples (n =101) were collected from throughout the lower South Island of New Zealand, with a maximum distance between samples of approximately 265 km (Fig. 1). All samples belonged to the Southern lineage of rock wren identified by Weston and Robertson (2015). Sampling was undertaken from 2009 to 2012 during the austral summer breeding season. Birds were initially attracted using audio playback. Due to their limited flight capability (Higgins et al. 2001), they were then either caught in hand nets or ‘herded’ into mist nets. The capture location of each individual was recorded using a Garmin handheld GPS. Resampling of birds was prevented by tagging each bird with a single metal leg band issued by the New Zealand Department of Conservation as part of the national bird banding scheme. Catching effort focused on adult pairs to avoid sampling relatives. Blood samples (<20 μl) were obtained by brachial vein puncture with a sterile needle (25G × 5/8”) and collection of the blood with a glass capillary tube for immediate transfer into 1 ml of Queen’s lysis buffer (10 mMTris, 10 mMNaCl, 10 mM Na-EDTA, 1 % n-lauroylsarcosine; pH 7.5) (Seutin et al. 1991). DNA was extracted and purified from the whole blood using a standard 5 % Chelex protocol (Walsh et al. 1991).

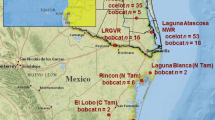
Sampling locations of 101 rock wren (Xenicus gilviventris) throughout the mountainous regions of the lower South Island. Sampling locations are labelled: Hr Haast Range n = 17; C Lake Crucible n=4; Ma Matukituki n = 6; A Lake Adelaide n = 6; H Upper Hollyford n = 25, Mn Lake Marion n = 2; Mk Monkey Creek n = 4; M Murchison Mtns n = 14; E Eyre Mtns n = 2; R Lake Roe n = 16; Mc Lake MacArthur n = 5
DNA sexing
Although rock wren are sexually dimorphic (Higgins et al. 2001), differences can be subtle and vary throughout their range (pers. obs). Therefore, the sex of each bird was reaffirmed using a DNA test. In birds, females are heterogametic, i.e. have two different sex chromosomes (ZW) and males are homogametic (ZZ). Consequently, detection of the female-specific W chromosome differentiates the sexes (Ellegren and Sheldon 1997; Dubiec and Zagalska-Neubauer 2006). We used the 2550F/2718R primer pair developed to amplify sex-specific versions of introns in the chromo-helicase-DNA binding protein (CHD) (Fridolfsson and Ellegren 1999). PCRs were performed in a 10 µl reaction containing 10–30 ng of template DNA, 1 × PCR buffer, 1.5mM of MgCl2, 200 µM of each dNTP, 0.5 unit of Taq polymerase (Bioline USA, Inc, Randolph MA 02368–4800) and 0.5 pmol of each primer (Robertson and Stephenson 2008). The thermal cycling parameters were an initial three minute denaturation at 94 °C, followed by a touchdown of 10 cycles at 94°C/15 s, 55→45°C/25 s (−1 °C per cycle) and 72°C/35 s, then 25 cycles at 94°C/15 s, 45°C/25 s and 72°C/35 s. PCR products were separated in 2 % agarose gels, run in standard TAE buffer, and visualized using SYBR® Safe DNA Gel Stain (Life Technologies).
Microsatellite genotyping
All 101 samples were genotyped at 13 microsatellite loci (Xgilv 1, Xgilv 4, Xgilv 8, Xgilv 9, Xgilv 12, Xgilv 16, Xgilv 19, Xgilv 20, Xgilv 21, Xgilv 23, Xgilv 27, Xgilv 28, Xgilv 31) previously developed for rock wren using 454 next generation sequencing (Weston and Robertson 2014). Electrophoresis of amplified PCR products was performed using an ABI 3730xl Genetic Analyser (Applied Biosystems) with GeneScanTM 500 LIZsize standard and scored using GENEMAPPER version 4.0 (Applied Biosystems). Tests for Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were conducted in GENEPOP version 4.1 (Rousset 2008) employing the Markov Chain method with 10,000 dememorisations, 1,000 batches and 10,000 iterations. Significance levels were adjusted for multiple comparisons among loci following standard Bonferroni correction (Rice 1989).
To check for genotyping error, 10 % of all samples were run twice for each locus. The error rate per allele was calculated by dividing the number of mismatched alleles by the total number of repeat-genotyped alleles (Hoffman and Amos 2005).
Population-level genetic diversity and differentiation
Levels of genetic diversity were evaluated for each of the four sampling locations where n > 10 (i.e. Haast Range, Upper Hollyford, Murchison Mountains and Lake Roe). Mean number of alleles per locus and observed/expected heterozygosity (Ho/He) were calculated using GenAlEx v.6.41 (Peakall and Smouse 2006). Allelic richness, a measure of allelic diversity that accounts for differences in sample size, and the inbreeding coefficient FIS were calculated in FSTAT v.2.9.3 (Goudet 2001). The significance of FIS values was calculated based on a significant deviation from HWE (i.e. heterozygote deficiency for positive values) over 1300 randomisations (Goudet 2001). Weir and Cockerham’s (1984) pairwise F ST values were estimated to evaluate the degree of genetic differentiation among sampling locations. Significance was tested over 10,000 permutations and strict Bonferroni corrections for multiple comparisons among loci were applied (Rice 1989). For comparison, genetic differentiation between each pair of sampling locations was also estimated using the statistic Jost’s D (Jost 2008) calculated in SMOGD v.1.2.5 (Crawford 2010).
Migration rates
To assess geneflow among sampling locations with >10 individuals, recent migration rates (over the last 2–3 generations) were estimated using Bayesian inference implemented in BayesAss 3.0 (Wilson and Rannala 2003). Rates are provided as the proportion of individuals in the receiving population that are migrants derived from the source population, per generation. The programme was run with a Bayesian Markov Chain Monte Carlo (MCMC) length of 10,000,000 iterations, a burn-in period of 2000,000 iterations and sampling every 100 generations. Various mixing parameter values were trialled to achieve optimal acceptance rates of 20–60 % for migration rates (m), allele frequency (a) and inbreeding coefficients (f); in the final model parameter values were set at: m = 0.3, a = 0.5 and f = 0.6. Convergence was confirmed through the assessment of the plotted log-probability using Tracer 1.5 (Rambaut and Drummond 2007) and by comparing the posterior mean parameter estimates amongst multiple runs for concordance.
Spatial genetic structure
To test for a positive correlation between genetic relatedness and linear (Euclidian) geographic distance (isolation by distance or ‘IBD’), Mantel tests were performed using coefficients of genetic relatedness (Queller and Goodnight 1989) between each pair of the 101 individuals sampled. Significance of correlations was estimated over 9999 replicates using GenAlEx v.6.41 (Peakall and Smouse 2006).
We also tested for spatial autocorrelation, the correlation of genotypes between pairs of individuals separated by similar geographic distances or which fall into specified ‘distance classes’, using a multivariate approach in GenAlEx v.6.41 (Peakall and Smouse 2006). Twelve arbitrary distance classes were constructed to contain approximately equal numbers of samples. A correlogram is produced showing the autocorrelation coefficient r as a function of distance class. Where fine-scale structure exists, r is typically greater than zero for shorter distance classes and less than zero for larger distance classes. The lowest distance at which the correlogram crosses the x-axis may be interpreted as the extent of non-random (positive) genetic structure or ‘genetic patch size’ i.e., the distance at which samples become genetically independent (Smouse and Peakall 1999). However, the interpretation of the x-intercept in this regard needs to consider the interplay of the distance class sizes chosen, sample sizes for each distance class and the true (but unknown) extent of genetic structure (Smouse and Peakall 1999). For this reason, we experimented with different distance class intervals to ensure approximately equal but adequate sample sizes and that our sampling intervals were not greater than the scale of genetic structure.
Two methods are used to test the significance of the autocorrelation in GenAlEx: random permutation and bootstrap estimates of r. Random permutations (9,999) are used to generate a distribution of permuted (rp) values under the assumption of no spatial structure, from which a 95 % null confidence interval is calculated. If the observed r value falls outside of this confidence interval, then significant spatial genetic structure can be inferred. Bootstrapping is used to place a confidence interval around each observed estimate of r. The bootstrap autocorrelation coefficient (r bs) is calculated for each of 9999 bootstraps and when the estimated bootstrap confidence interval does not straddle r = 0, significant spatial structure is inferred. The overall significance of spatial autocorrelation across all distance classes is also tested over 9,999 permutations under the null hypothesis that the entire correlogram is ‘flat’ against the alternative hypothesis that it is not. Nonparametric heterogeneity tests were also performed to test for significant differences in spatial genetic structure patterns between the sexes (Smouse et al. 2008). The tests are performed at each distance class where the null hypothesis predicts overlap in the 95 % CIs between the sexes and also at the whole correlogram level under the null hypothesis that the correlograms from both sexes are homogeneous.
Mean pairwise relatedness among individuals within each sampling location was also calculated in GenAlEx using the Queller and Goodnight (1989) index of relatedness (r). The overall index ranges between −1 and 1, with values of r approximating 0.5 for parent-offspring or full-siblings, c. 0.25 for half-siblings and zero for unrelated individuals (Queller and Goodnight 1989). The 95 % confidence intervals about the mean values were estimated using 10,000 bootstraps.
To further investigate genetic structure and the geographic location of any genetic discontinuities within the lower South Island, we utilized two Bayesian clustering methods implemented in Geneland (Guillot et al. 2005) and STRUCTURE v.2.3.3 (Pritchard et al. 2000). Both methods assign individuals probabilistically to genetic clusters (K) by calculating membership coefficients per individual per cluster (Q). Geneland uses individual GPS coordinates as prior information, whilst for STRUCTURE, the sampling location was provided as prior spatial information. In both cases, an admixture model was used and uncorrelated allele frequencies, as this is less prone to algorithm instabilities and departure from model assumptions (e.g. presence of isolation-by-distance) (Guillot 2012). We tested for K = 1–7 (seven being the number of broad geographic regions sampled) with ten independent runs performed for each value of K.
In Geneland, after 1,000,000 Markov Chain Monte Carlo (MCMC) iterations, post processing was carried out on the run with the highest average posterior probability to compute the posterior probability of population membership for every pixel in the spatial domain (150 × 250 pixels, 2,000 iterations discarded as burn-in). For STRUCTURE, results are based on 1,000,000 MCMC iterations following a burn-in of 100,000. The true value of K was inferred by evaluating Q values, the mean log-likelihood of the data (ln P(X|K)) and using the Delta-K (∆K) method (Evanno et al. 2005) implemented in Structure Harvester (Earl and vonHoldt 2012). Results were averaged across the ten runs using Clumpp v.1.1.2 (Jakobsson and Rosenberg 2007) and visualized using Distruct v.1.1 (Rosenberg 2004).
Tests for sex-biased dispersal
We conducted five independent tests for sex-biased dispersal using FSTAT v.2.9.3 (Goudet 2001). Firstly, we compared values of Weir and Cockerham’s (1984) F ST estimator θST among populations for males and females. It is expected that the dispersing sex will have a lower θST given that dispersal homogenises allelic frequencies. Secondly, we examined differences in the inbreeding coefficient (FIS) between the sexes. The dispersing sex should display a higher FIS than the more philopatric sex indicative of greater genetic structuring among sub populations (Goudet et al. 2002). This underlying genetic structure leads to a heterozygote deficit known as the ‘Wahlund effect’ and produces an effect analogous to inbreeding (Goudet et al. 2002). This is because individuals sampled from a single location will be a mixture of both residents and immigrants in the dispersing sex, meaning that homozygotes will be more frequent and heterozygotes less frequent than expected based on the calculated allele frequency for the combined population. FSTAT v.2.9.3 also calculates Hamilton’s (1971) index of relatedness r = 2F ST /(1 + FIT), using an estimator strictly equivalent to Queller and Goodnight’s (1989). Relatedness amongst members of the dispersing sex is predicted to be lower (Goudet et al. 2002). For the last two tests, we calculated the Assignment Index statistic (AI) and tested for differences in: (1) the mean (mAIc), and; (2) variance (vAIc) between the sexes. AIc values are centred on zero and calculate the probability for each genotype to be represented in the sampled population. Resident individuals are likely to have genotypes with an above-average likelihood of occurring in the sample, which should lead to positive AIc values. Consequently, mAIc should be higher in the philopatric sex and vAlc should be higher in the dispersing sex because of the inclusion of both immigrant and resident genotypes (Lampert et al. 2003; Helfer et al. 2012).
Results
The mean genotyping error rate across all 13 loci was low, with 0.0116 alleles resulting in an allelic mismatch. There was no evidence for linkage disequilibrium between any pair of loci, or significant consistent departure from HWE for any locus after significance levels were adjusted for multiple pairwise comparisons using the sequential Bonferroni correction.
Population-level genetic diversity and differentiation
Levels of genetic diversity were similar among the four sampling locations (He 0.524–0.557), however the Haast Range exhibited notably lower levels of allelic diversity (Table 1). Values of FIS were positive for all sampling locations, indicative of inbreeding, with the highest values for the Haast Range and Murchison Mountains, though this value was only significantly different from HWE for the Haast Range (Table 1). Levels of genetic differentiation among sampling locations were comparable among the two distance methods (F ST and Jost’s D), both showing the greatest differentiation among the Haast Range and all other sampling locations (Table 2).
Migration rates among sampling locations
The mean posterior probabilities of migration rates showed that the majority of individuals originated from the location within which they were sampled, with the Haast Range having the highest proportion of residents (m = 0.94) (Table 3). The Murchison Mountains was the only significant source of migrants for all locations (m = 0.15–0.27), and migration was asymmetrical with almost no geneflow into the Murchison Mountains from any other sampling location (Table 3).
Spatial genetic structure
Across the lower South Island, a weak but significant IBD relationship of decreasing pairwise relatedness over increasing geographic distance was detected (R2 = 0.080, P < 0.001) (see Supplementary Information). Spatial autocorrelation was highly significant (P < 0.001) overall, indicating that rock wren are non-randomly distributed across the landscape (Fig. 2). Autocorrelation was positive and significant (P < 0.001)for the first six distance classes thus providing evidence that individuals <50 km apart are on average more genetically similar than spatially random pairs. The correlogram crosses the X-axis at c. 70 km, indicating that individuals separated by >70 km are less genetically similar than expected at random (Fig. 2).

Correlogram showing autocorrelation across distance classes for 101 rock wren (Xenicus gilviventris) sampled throughout the lower South Island. The endpoint of each distance class is labelled on the x-axis. Error bars represent bootstrapped 95 % confidence intervals about mean r values. Dashed lines represent upper (U) and lower (L) 95 % confidence limits under the null hypotheses that genotypes are randomly distributed across the landscape
The heterogeneity tests revealed significant overall differences among the correlograms of males and females (P = 0.01). Males exhibited higher positive autocorrelation than females up to distances of 25 km (Fig. 3). However, these differences were only significant at the 1–2 km and 5–10 km distance classes (P < 0.05). Beyond 25 km, the pattern generally reversed with males exhibiting lower levels of genetic similarity than females, though differences were only significant at the 70–100 km distance class (P < 0.05).

Correlogram comparing auto correlation across distance classes for male (n = 58) and female (n = 43) rock wren (Xenicus gilviventris) sampled throughout the lower South Island. The endpoint of each distance class is labelled on the x-axis. Error bars represent bootstrapped 95 % confidence intervals about mean r values
Levels of inter-individual relatedness differed significantly among sampling locations (Fig. 4). Individuals from the Haast Range, Murchison Mountains and Lake Roe all exhibited greater levels of relatedness (mean 0.090 – 0.111) than individuals within the Upper Hollyford (mean = 0.025, 95 % CI −0.002 to 0.029).

Mean pairwise genetic relatedness estimates from four rock wren (Xenicus gilviventris) sampling locations in the lower South Island. Error bars are the 95 % CI as determined by bootstrapping. Upper Hollyford n = 25; Haast Range n = 17; Murchison Mountains n = 14; Lake Roe n = 16
The Bayesian cluster analysis using both Geneland and STRUCTURE showed strongest support for two genetic populations (K = 2) (see Supplementary Information).
Cluster assignments corresponded with spatial locations (Fig. 5). Cluster 1 (green) encompassed all individuals sampled from the Haast Range and Mount Aspiring National park (i.e. Lake Crucible and the Matukituki); Cluster 2 (white) encompassed all other sampling locations in the lower South Island. Individual assignment probabilities to each cluster were high using Geneland (0.99 on average); though lower in STRUCTURE (0.92 and 0.84 to clusters 1 and 2, respectively).

Estimated rock wren (Xenicus gilviventris) membership (n = 101) to two genetic clusters as generated by a STRUCTURE and b Geneland. Each vertical bar in a represents a single individual and individuals are ordered by sampling location from north to south according to latitude. For Geneland b, black dots on map denote sampling locations. Sampling locations used in population-level analysis (i.e. where n > 10) are labelled: Hr Haast Range, H Upper Hollyford, M Murchison Mountains, R Lake Roe
Sex-biased dispersal
Results from all five tests for sex-biased dispersal were not significant (P > 0.05) (Table 4). Two of the tests indicated a female-bias dispersal system (females: lower θST, lower relatedness (r)). However the assignment indices (mAIc, vAIc) supported a male-bias (males: lower mean assignment index and higher variance in assignment index).
Discussion
We evaluated the genetic structure of rock wren over spatial scales ranging up to c. 250 km and detected a large genetic patch size for rock wren (c. 70 km). Among sampling locations, we found significant differences in levels of inter-individual relatedness and asymmetrical gene flow, indicative of source–sink dynamics. Cluster analysis showed strongest support for two genetic populations; one comprising all individuals sampled from the Haast Range and Mount Aspiring National Park (i.e. Lake Crucible and the Matukituki), the other, encompassing all other sampling locations in the lower South Island.
Population-level inferences and spatial genetic structure
Levels of genetic diversity were similar among the four sampling locations, though low levels of allelic diversity together with high levels of inbreeding and relatedness were detected for the Haast Range. It is unlikely that family groups were unintentionally included during sampling of the Haast Range thereby inflating relatedness, because this location was sampled late in the season—i.e. end of February—early March, by which time most juveniles are likely to have dispersed (Michelsen-Heath 1989). Further, all individuals sampled were identified as adults (seven females, ten males) in the same manner as the other sampling locations. Juvenile rock wren are distinguishable (albeit subtly) as they do not moult to adult plumage until c. 3 months post-fledging, by which time they have typically paired and established territories of their own (Michelsen-Heath 1989). Another explanation for the high inbreeding in this area could be its peripheral location, right at the western-most extent of their range (Fig. 1). Being peripheral, the Haast Range naturally has fewer neighbours for genetic exchange than do core populations (Segelbacher and Storch 2002). Landscape connectivity has been shown to be closely linked to the amount of genetic diversity in small and isolated populations (Trizio et al. 2005; Keyghobadi 2007) and peripheral populations often exhibit low genetic diversity and greater genetic differentiation (Eckert et al. 2008). Cluster analysis did however group individuals from the Haast Range and Mount Aspiring National Park (i.e. Lake Crucible and the Matukituki) together indicating high gene flow among these areas, which are located a maximum of c. 40 km from each other. It should also be acknowledged that the variation at neutral microsatellite loci described in this study may not capture all of the genetic diversity present within southern rock wren populations (Kirk and Freeland 2011; Grueber et al. 2015).
A genetic discontinuity was identified between theHaast Range/Mount Aspiring area and the Upper Hollyford. It is possible that this discontinuity is associated with a lack of landscape connectivity between the Matukituki Valley and the Upper Hollyford, with Lake Wakatipu and several major river systems in the area forming a barrier to gene flow. This discontinuity is also reflected in the migration rates among sampling locations, with most gene flow detected among the three southern-most areas; the Upper Hollyford, Murchison Mountains and Lake Roe.
It is important to consider that in this study, we applied Euclidian (straight-line) distances between individuals/sampling locations. Another approach would be to calculate the ‘effective distance’ using landscape genetic methods such as least-cost distance, which measures the path between two locations, avoids barriers, and minimises the ‘resistance’ of the matrix habitat (Adriaensen et al. 2003). This method would provide an interesting comparison for rock wren, given that they are an alpine species only very rarely recorded below the bushline. The effective distance rock wren travel above the bushline and along ridges may differ substantially from the Euclidian distance conventionally used in tests of isolation by distance and spatial autocorrelation.
A genetic patch size of c. 70 km for rock wren initially seems unexpectedly large, especially given their poor flight ability (Higgins et al. 2001), small territory size (c. 0.6–4.2 hectares; Michelsen-Heath 1989) and previous observations indicating that the majority of juveniles disperse to within 500 m of their place of fledging (Michelsen-Heath 1989). However, given that rock wren demonstrate a pattern of isolation by distance suggesting a ‘stepping stone’ model of geneflow among populations (Kimura and Weiss 1964), genes can be dispersed over distances far greater than rock wren dispersal ability would predict, as has been shown for other sedentary alpine bird species (Segelbacher and Storch 2002; Fedy et al. 2008). Further, although a majority (75 %) of juvenile birds were observed dispersing short distances by Michelsen-Heath (1989), the remaining 25 % were not resighted. Therefore, it is possible that at least some of these birds dispersed over much greater distances.
Spatial autocorrelation has not often been used to assess avian fine-scale genetic structure and where it has been applied, genetic patch size has been found to be highly variable in passerines(<200 m–c.15 km) and strongly influenced by social system, demography and landscape features (Double et al. 2005; Beck et al. 2008; Wilson et al. 2011; Nelson-Flower et al. 2012; Liebgold et al. 2013; Vangestel et al. 2013). We might expect long-distance dispersal to be selected for in a patchy and dynamic environment such as the alpine zone in order for birds to locate and colonise new habitat and for populations to persist (Johst et al. 2002). This could also partly explain the relatively large genetic patch size in rock wren, compared to forest/woodland species for example, where resources and habitat may be more homogenous (Peakall et al. 2003; Beck et al. 2008; Vangestel et al. 2013). However, given that the probability of successful dispersal within a harsh and unpredictable environment is likely to be lower, remaining in the natal area and inheriting a breeding site of proven quality can also be expected to constitute an evolutionarily stable strategy (Ronce 2007). There is some evidence indicating that non-random mating is contributing to the fine-scale population structure observed in rock wren with higher positive spatial genetic autocorrelation among males over shorter distances (significantly from 1 – 10 km); combined with a higher F ST and higher relatedness for males within populations. Whilst results for the latter tests were indicative, they were not significant, though these methods are known to have limited power unless the bias in dispersal is extreme (e.g. an 80:20 sex ratio of dispersers; (Goudet et al. 2002). The fact that the difference in positive autocorrelation between the sexes declined over larger distances in rock wren is interesting, though not entirely unexpected given growing evidence suggesting that selective pressures (e.g. access to food and mates) affect males and females differentially, resulting in sex-specific and often antagonistic dispersal strategies over different spatial scales (Jost 2008; Yannic et al. 2012; Vangestel et al. 2013).
Management implications
We focused this study within the lower South Island due to the higher sampling coverage in this area, and because conservation management is already underway in this southern region. With further sampling, this approach could also be applied to other areas, for example within the north of the rock wren’s range where differences in habitat and other environmental factors may differentially affect gene flow. Understanding the spatial scale of gene flow in rock wren has several important implications for management.
Firstly, patterns of gene flow among the southern sites are suggestive of source–sink dynamics. The Murchison Mountains appear to be a particularly important source of migrants for other locations. This area is already under intensive conservation management as it constitutes a Special Area reserved for the endangered Takahe (Porphyrio hochstetteri). Since the discovery of the Takahe in 1948, the Special Area has been managed to control predators and deer in an effort to improve habitat for the birds (Parkes et al. 1978; Wickes et al. 2009). These management practices are also likely to benefit rock wren by boosting reproductive success. Conversely, for the Upper Hollyford and Lake Roe, most dispersal is occurring into, but not out of these areas. Further, the lack of pairwise relatedness among individuals within the Upper Hollyford is also indicative of a sink (King et al. 2000). Low relatedness can occur in a sink population if a large proportion of recruitment results from immigration and fewer close relatives are present. In contrast, higher relatedness should be detected in closed and self-sustaining populations (Peery et al. 2008). Sinks typically occur in poor quality habitats, where productivity is insufficient to balance mortality (Pulliam 1988; Dias 1996). Consequently, without migration from outside sources, sink populations would be expected to eventually become extinct (Pulliam 1988). High rates of predation by introduced mustelids reported for rock wren from these areas may be contributing to their demographic instability (Department of Conservation, unpublished data). Anthropogenic-induced mortality, such as predation by introduced species, has been previously implicated in source–sink dynamics (Woodford and McIntosh 2010; Balogh et al. 2011). Demographic field data on primary population parameters (productivity, mortality, recruitment) from different rock wren populations are now required to further examine population dynamics, particularly under different predator management regimes.
There is evidence for significant genetic differentiation north and south of Lake Wakatipu. This may reflect impaired connectivity in this area due to natural landscape barriers such the lake and several major rivers. Alternatively, or synergistically; there may be anthropogenic factors in force such as high levels of predation on dispersing individuals. In the case of the latter, predator-controlled areas functioning as ‘habitat corridors’ could be an option to increase connectivity (Mech and Hallett 2001). Although lower allelic diversity and relatively higher levels of inbreeding were detected within rock wren from the Haast Range, this is also the most genetically differentiated of the Southern areas. On this basis, the Haast Range represents an important population in terms of conserving the among-population component of genetic variation present within southern rock wren.
Lastly, climate change is predicted to have a disproportionate impact on alpine specialists, as populations become increasingly isolated atop shrinking alpine islands (Galbreath et al. 2009; Dirnböck et al. 2011; Rubidge et al. 2012). Our findings indicate that conserving natural connectivity among rock wren populations across relatively large spatial scales is essential in maintaining not only metapopulation dynamics and the adaptive genetic diversity of populations, but also to facilitate migration of individuals southwards into relatively cooler climes (Heller and Zavaleta 2009; Moritz and Agudo 2013).
References
Abbott CL, Põldmaa T, Lougheed S, Clarke M, Boag PT (2002) Hierarchical analysis of genetic population structure in the noisy miner using DNA microsatellite markers. Condor 104:652–656
Adriaensen F, Chardon JP, De Blust G, Swinnen E, Villalba S, Gulinck H, Matthysen E (2003) The application of ‘least-cost’ modelling as a functional landscape model. Landsc Urban Plan 64:233–247
Andreasen AM, Stewart KM, Longland WS, Beckmann JP, Forister ML (2012) Identification of source–sink dynamics in mountain lions of the Great Basin. Mol Ecol 21:5689–5701
Baker AJ, Daugherty CH, Colbourne R, McLennan JL (1995) Flightless brown kiwis of New Zealand possess extremely subdivided population structure and cryptic species like small mammals. Proc Natl Acad Sci USA 92:8254–8258
Balogh AL, Ryder TB, Marra PP (2011) Population demography of Gray Catbirds in the suburban matrix: sources, sinks and domestic cats. J Ornithol 152:717–726
Bech N, Boissier J, Drovetski S, Novoa C (2009) Population genetic structure of rock ptarmigan in the ‘sky islands’ of French Pyrenees: implications for conservation. Anim Conserv 12:138–146
Beck NR, Peakall R, Heinsohn R (2008) Social constraint and an absence of sex-biased dispersal drive fine-scale genetic structure in white-winged choughs. Mol Ecol 17:4346–4358
Caizergues A, Bernard-Laurent A, Brenot JF, Ellison L, Rasplus JY (2003) Population genetic structure of rock ptarmigan Lagopus mutus in Northern and Western Europe. Mol Ecol 12:2267–2274
Crawford NG (2010) SMOGD: software for the measurement of genetic diversity. Mol Ecol Resour 10:556–557
Crochet PA (2000) Genetic structure of avian populations - Allozymes revisited. Mol Ecol 9:1463–1469
Dias PC (1996) Sources and sinks in population biology. Trends Ecol Evol 11:326–330
Dirnböck T, Essl F, Rabitsch W (2011) Disproportional risk for habitat loss of high-altitude endemic species under climate change. Glob Change Biol 17:990–996
Double MC, Peakall R, Beck NR, Cockburn A (2005) Dispersal, philopatry, and infidelity: dissecting local genetic structure in superb fairy-wrens (Malurus cyaneus). Evolution 59:625–635
Dubiec A, Zagalska-Neubauer M (2006) Molecular techniques for sex identification in birds. Biol Lett 43:3–12
Earl D, vonHoldt B (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Eckert CG, Samis KE, Lougheed SC (2008) Genetic variation across species’ geographical ranges: the central–marginal hypothesis and beyond. Mol Ecol 17:1170–1188
Ellegren H, Sheldon BC (1997) New tools for sex identification and the study of sex allocation in birds. Trends Ecol Evol 12:255–259
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fedy BC, Martin K, Ritland C, Young J (2008) Genetic and ecological data provide incongruent interpretations of population structure and dispersal in naturally subdivided populations of white-tailed ptarmigan (Lagopus leucura). Mol Ecol 17:1905–1917
Fridolfsson AK, Ellegren H (1999) A simple and universal method for molecular sexing of non-ratite birds. J Avian Biol 30:116–121
Funk WC, McKay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. Trends Ecol Evol 27:489–496
Furrer RD, Pasinelli G (2015) Empirical evidence for source–sink populations: a review on occurrence, assessments and implications. Biol Rev. doi:10.1111/brv.12195
Galbreath KE, Hafner DJ, Zamudio KR (2009) When cold is better: climate-driven elevation shifts yield complex patterns of diversification and demography in an alpine specialist (american pika, ochotona princeps). Evolution 63:2848–2863
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (Version 2.9.3). Available at http://www2.unil.ch/popgen/softwares/fstat.html
Goudet J, Perrin N, Waser P (2002) Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol Ecol 11:1103–1114
Greenwood PJ (1980) Mating systems, philopatry and dispersal in birds and mammals. Anim Behav 28:1140–1162
Grueber C, Knafler G, King T, Senior A, Grosser S, Robertson B, Weston K, Brekke P, Harris CW, Jamieson I (2015) Toll-like receptor diversity in 10 threatened bird species: relationship with microsatellite heterozygosity. Conserv Genet 16:595–611
Guillot G (2012) Population genetic and morphometric data analysis using R and the GENELAND program, User Manual
Guillot G, Mortier F, Estoup A (2005) GENELAND: a computer package for landscape genetics. Mol Ecol Notes 5:712–715
Hamilton WD (1971) Selection of selfish and altruistic behaviour in some extreme models. In:JF Eisenberg, Dilon WS (eds) Man and beast: comparative social behavior. Smithsonian Institute Press, Washington, D.C. pp 57–91
Helfer V, Broquet T, Fumagalli L (2012) Sex-specific estimates of dispersal show female philopatry and male dispersal in a promiscuous amphibian, the alpine salamander (Salamandra atra). Mol Ecol 21:4706–4720
Heller NE, Zavaleta ES (2009) Biodiversity management in the face of climate change: a review of 22 years of recommendations. Biol Conserv 142:14–32
Higgins PJ, Peter JM, Steele WK (eds) (2001) Handbook of Australian. Oxford University Press, Melbourne, New Zealand and Antarctic Birds
Hoffman JI, Amos W (2005) Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion. Mol Ecol 14:599–612
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Johst K, Brandl R, Eber S (2002) Metapopulation persistence in dynamic landscapes: the role of dispersal distance. Oikos 98:263–270
Jost LOU (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Keyghobadi N (2007) The genetic implications of habitat fragmentation for animals. Can J Zool 85:1049–1064
Kimura M, Weiss GH (1964) The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49:561–576
King TM, Williams M, Lambert DM (2000) Dams, ducks and DNA: identifying the effects of a hydro-electric scheme on New Zealand’s endangered blue duck. Conserv Genet 1:103–113
Kirk H, Freeland JR (2011) Applications and implications of neutral versus non-neutral markers in molecular ecology. Int J Mol Sci 12:3966–3988
Lampert KP, Rand AS, Mueller UG, Ryan MJ (2003) Fine-scale genetic pattern and evidence for sex-biased dispersal in the túngara frog, Physalaemus pustulosus. Mol Ecol 12:3325–3334
Liebgold EB, Gerlach NM, Ketterson ED (2013) Similarity in temporal variation in sex-biased dispersal over short and long distances in the dark-eyed junco, Junco hyemalis. Mol Ecol 22:5548–5560
Mech SG, Hallett JG (2001) Evaluating the effectiveness of corridors: a genetic approach. Conserv Biol 15:467–474
Michelsen S (1982) Habitat requirements and feeding behaviour of the rock wren, Xenicus gilviventris in Mount Cook National Park, New Zealand. Post-Grad. Dip. Wildlife Management thesis, University of Otago
Michelsen-Heath S (1989) The breeding biology of the rock wren, Xenicus gilviventris, in the Murchison Mountains, Fiordland National Park, New Zealand. MSc thesis, University of Otago
Moritz C, Agudo R (2013) The future of species under climate change: resilience or decline? Science 341:504–508
Nelson-Flower MJ, Hockey PAR, O’Ryan C, Ridley AR (2012) Inbreeding avoidance mechanisms: dispersal dynamics in cooperatively breeding southern pied babblers. J Anim Ecol 81:876–883
O’Keefe K, Ramakrishnan U, Van Tuinen M, Hadly EA (2009) Source–sink dynamics structure a common montane mammal. Mol Ecol 18:4775–4789
Parkes J, Tustin K, Stanley L (1978) The history and control of red deer in the Takahe area, Murchison Mountains, Fiordland National Park. New Zealand Journal of Ecology 1:145–152
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Peakall R, Ruibal M, Lindenmayer DB (2003) Spatial autocorrelation analysis offers new insights into gene flow in the Australian bush rat, Rattus fuscipes. Evolution 57:1182–1195
Peery MZ, Beissinger SR, House RF, Bérubé M, Hall LA, Sellas A, Palsbøll PJ (2008) Characterizing source sink dynamics with genetic parentage assignments. Ecology 89:2746–2759
Perrin N, Mazalov V (2000) Local competition, inbreeding, and the evolution of sex-biased dispersal. Am Nat 155:116–127
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pulliam HR (1988) Sources, sinks and population regulation. Am Nat 132:652–661
Pusey AE (1987) Sex-biased dispersal and inbreeding avoidance in birds and mammals. Trends Ecol Evol 2:295–299
Queller DC, Goodnight KF (1989) Estimating relatedness using genetic markers. Evolution 43:258–275
Rambaut A, Drummond AJ (2007) Tracer v 1.5. Available from http://beast.bio.ed.ac.uk/Tracer
Rice WR (1989) Analysing tables of statistical tests. Evolution 43:223–225
Robertson BC, Stephenson BM (2008) DNA sexing of the critically endangered New Zealand storm petrel (Oceanites maorianus, or Pealeornis maoriana). Notornis 55:209–211
Ronce O (2007) How does if feel to be a rolling stone? Ten questions about dispersal evolution. Annu Rev Ecol Syst 38:231–253
Rosenberg NA (2004) DISTRUCT: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8:103–106
Rubidge EM, Patton JL, Lim M, Burton AC, Brashares JS, Moritz C (2012) Climate-induced range contraction drives genetic erosion in an alpine mammal. Nat Clim Change 2:285–288
Segelbacher G, Storch I (2002) Capercaillie in the Alps: genetic evidence of metapopulation structure and population decline. Mol Ecol 11:1669–1677
Seutin G, White BN, Boag PT (1991) Preservation of avian blood and tissue samples for DNA analyses. Can J Zool 69:82–90
Smouse PE, Peakall R (1999) Spatial autocorrelation analysis of individual multiallele and multilocus genetic structure. Heredity 82:561–573
Smouse PE, Peakall R, Gonzales E (2008) A heterogeneity test for fine-scale genetic structure. Mol Ecol 17:3389–3400
Trizio I, Crestanello B, Galbusera P, Wauters LA, Tosi G, Matthysen E, Hauffe HC (2005) Geographical distance and physical barriers shape the genetic structure of Eurasian red squirrels (Sciurus vulgaris) in the Italian Alps. Mol Ecol 14:469–481
Vangestel C, Callens T, Vandomme V, Lens L (2013) Sex-Biased Dispersal at Different Geographical Scales in a Cooperative Breeder from Fragmented Rainforest. PLoS ONE, 8
Walsh PS, Metzger DA, Higuchi R (1991) Chelex® 100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques 10:506–513
Waples RS, Gaggiotti O (2006) What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Weston K, Robertson B (2014) Isolation and characterisation of 14 microsatellites for the New Zealand rock wren Xenicus gilviventris. Conserv Genet Resour 6:115–117
Weston KA, Robertson BC (2015) Population structure within an alpine archipelago: strong signature of past climate change in the New Zealand rock wren (Xenicus gilviventris). Mol Ecol 24:4778–4794
Wickes C, Crouchley D, Maxwell J (2009) Takahe (Porphyrio hochstetteri) recovery plan 2007–2012. Department of Conservation, Wellington
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Wilson AG, Arcese P, Chan YL, Patten MA (2011) Micro-spatial genetic structure in song sparrows (Melospiza melodia). Conserv Genet 12:213–222
Woodford DJ, McIntosh AR (2010) Evidence of source–sink metapopulations in a vulnerable native galaxiid fish driven by introduced trout. Ecol Appl 20:967–977
Yannic G, Basset P, Büchi L, Hausser J, Broquet T (2012) Scale-specific sex-biased dispersal in the valais shrew unveiled by genetic variation on the y chromosome, autosomes, and mitochondrial dna. Evolution 66:1737–1750
Acknowledgments
We thank all those who contributed to the sampling of rock wren in the field. Bruce Robertson especially thanks the Robertson family for their help within the Upper Hollyford Valley. Sabrina Taylor thanks Nick Torr for the use of his truck, his great company and help with rock wren sampling in the Upper Hollyford Valley. Fieldwork was conducted under the approval of the University of Otago Ethics Committee (No. 26/09) and DoC (Permits No. 2010/003; SO-27009-FAU; SO-24901-FAU; OT-26308-FAU). Project funding was gratefully received from the Mohua Charitable Trust, BirdLife International Community Conservation Fund, Brian Mason Scientific and Technical Trust, JS Watson Trust administered by Royal Forest and Bird Protection Society of New Zealand, Macpac Ltd. and the University of Otago.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Weston, K.A., Taylor, S.S. & Robertson, B.C. Identifying populations for management: fine-scale population structure in the New Zealand alpine rock wren (Xenicus gilviventris). Conserv Genet 17, 691–701 (2016). https://doi.org/10.1007/s10592-016-0815-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-016-0815-8