
Abstract
Phylogeographic patterns of Ammopiptanthus in northwestern China were examined with internal transcribed spacer (ITS) and three chloroplast intergenic spacers (trnH–psbA, trnL–trnF, and trnS–trnG). Two ITS genotypes (a–b) and 8 chloroplast haplotypes (A–H) were detected. Both ITS genotypes and chloroplast lineages were split in two geographic regions: western Xinjiang and the Alxa Desert. This lineage split was also supported by AMOVA analysis and the Mantel test. AMOVA showed that 89.81 % of variance in Ammopiptanthus occurred between the two geographic regions, and correlation between genetic distances and geographical distances was significant (r = 0.757, p < 0.0001). All populations in western Xinjiang shared haplotype A with high frequency, and range expansion was strongly supported by negative Fu’s FS value, and mismatch distribution analysis, whereas populations in the Alxa Desert had higher genetic diversity and structure. We speculate that the cold and dry climate during the early Quaternary fragmented habitats of the species, limiting gene flow between regions, and interglacial periods most likely led to the range expansion in western Xinjiang. The low genetic diversity of Ammopiptanthus indicate a significant extinction risk, and protective measures should be taken immediately.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phylogeographic studies can resolve unclear systematic classification problems of species (Avise 2000). Proper systematic classification is important for conservation of endangered species. Incorrect taxonomic classification will lead to ill-conceived conservation strategies. For example, species that may need protection could go unrecognized, or at the other end of the spectrum, and resources could be wasted on the organisms wrongly characterized and regarded as endangered (Frankham et al. 2002). Phylogeographic studies can also help in identifying intraspecific evolutionarily significant unit (ESU), a phylogenetic unit that is reciprocally monophyletic with its sister clade at a cytoplasmic locus and shows significant divergence of allele frequencies (Ryder 1986; Fraser and Bernatchez 2001). Recognizing ESUs within endangered species ensures that different lineages can be managed separately, and thus, high genetic diversity can be maintained (Crandall et al. 2000). In addition, phylogeography can provide other valuable genetic information, such as genetic diversity, population structure, and evolutionary history, which are essential to the development of broad-scale conservation strategies and planning of management actions for the conservation of the endangered species (Osborne et al. 2000; Browning et al. 2001; Moritz 1994).
The arid northwestern region of China comprises the entire Xinjiang Autonomous Region, the Hexi Corridor in Gansu Province, the Qaidam Basin in Qinghai Province, and western parts of the Helan Mountains in Inner Mongolia Autonomous Region (the Alxa Desert) (Dang et al. 2002). Many of the species in this area have fragmented populations and may be relicts of the Paleo-Mediterranean flora, such as Helianthemum songaricum, Gymnocarpos przewalskii, Reaumuria soongarica, Cynomorium songaricum; or the derivants of the Paleo-Mediterranean flora, such as Ammopiptanthus mongolicus (Tang 2000). This implies a close relationship among floras of the Mediterranean, Xinjiang, and Inner Mongolia region. The Hexi Corridor have acted as a conduit allowing plants to colonize these different regions.
Climate oscillations during the Quaternary have had profound effects on the phylogeographical patterns of species in the arid northwestern China (Su et al. 2011; Guo et al. 2010). Besides the climate oscillations, phylogeography of native species were apparently affected also by the aridization of interior Asia, partially driven by the Qinghai-Tibet Plateau (QTP) uplift and global cooling (Qin et al. 2013; Su and Zhang 2013). Cold and dry glacial periods have usually resulted in allopatric divergences in desert species, as well as a contraction and/or fragmentation of populations (Liu et al. 2012; Guo et al. 2010; Su et al. 2011; Wang et al. 2009; Jia et al. 2012); whereas warm interglacial periods have caused regional range expansions in some desert species (Guo et al. 2010; Li et al. 2012; Su and Zhang 2013).
Ammopiptanthus is an evergreen shrub and primarily occurs on sand dunes, gravel slopes, and terraces beside ravines that support an arid climate. It has multiple greyish-green branches, leathery leaves, yellow flowers (Cui 1998), and relies on insect pollination (Chen et al. 2009a, b). There has been long dispute about the classification of the genus Ammopiptanthus. Earlier classifications recognized two species, A. mongolicus and A.nunas, based on leaf morphology (Cui 1998). A. nunas mainly distributes in Ulugqat Country in western Xinjiang, and extend west to Kyrgyzstan, and A. mongolicus mainly distributes in the Alxa Desert in Inner Mongolia, and extend north to southern Mongolia (Cui 1998). However, more recently, taxonomists contended that the differences between the two species are of degree only, and the considerable variation in their morphology are caused by water availability, grazing, and season, similar with many other species of arid areas. Thus, Wei and Lock (2010) thought there should be only one species in Ammopiptanthus, A. mongolicus.
Because of insect pests (Bracon sp. and Nephopteryx sp.) and anthropogenic activities (e.g. coal mining, road construction, overgrazing and cutting for firewood) over the past several decades, Ammopiptanthus and its habitat is in decline. This drastic decline has warranted listing as endangered on the China Species Red List (Fu 1992). There have been a number of genetic or systematic studies about the Ammopiptanthus. Using inter-simple sequence repeat (ISSR) analysis, Ge et al. (2005) found moderate genetic differentiation in both A. mongolicus and A.nunas, and the two species are genetically distinct from each other, indicated by 63 % species-specific bands. Chen et al. (2009a, b) examined allozyme variation in A. nanus and A. mongolicus. They found low levels of genetic variation at population levels in both species, while moderate genetic differentiation existed among the A. nanus populations and significant genetic divergence among the A. mongolicus populations from the Tengger Desert and from Ulan Buh Desert/Helan Mountain. Based on nrITS sequences, Wang et al. (2006) found A. mongolicus and A.nunas were reciprocally monophyletic clades, whereas Xie and Yang (2012) got the reverse result. To clarify the classification and provide useful information for conservation in Ammopiptanthus, it is necessary to investigate the genetic structure and the phylogeographical pattern across the whole distribution range and use modern molecular techniques.
The chloroplast (cp) DNA of most plants is inherited from only one of the parents, typically maternal inheritance (Birky 2001; Avise 2000); thus, without the confounding effects of biparental inheritance, cpDNA lineages in natural populations can better trace the evolutionary history of species and display distinct geographical distributions (Avise 2000). However, cpDNA markers may only partly recover the phylogeographical history of a species (Mäder et al. 2010), thus additional use of biparentally inherited nuclear DNA markers might contribute more information to the evolutionary process (Liu et al. 2012). Recently, the combination of nuclear and chloroplast markers has been increasingly used in plant population genetic studies (Jia et al. 2012; Martins et al. 2011; Qin et al. 2013). In the present study, we used nuclear ITS sequence, and chloroplast markers including trnH–psbA, trnL–trnF, trnS–trnG, to investigate the full, range wide genetic variation of Ammopiptanthus to address the following questions: (1) do populations of Ammopiptanthus in western Xinjiang represent a species rank, A.nunas; (2) if so, what is genetic variation within and population differentiation between the two species; (3) were there similar range shifts within each of the two regions?
Materials and methods
Plant materials
A total of 392 individuals from 33 populations of the two species were sampled, covering almost the entire geographical range of its distribution. Populations 1–12 were sampled in western Xinjiang province (Region 1), and populations 13–33 were sampled in the Alxa Desert, including Inner Mongolia, Ningxia, and Gansu province (Region 2). Ten to 12 individuals were collected per population. Fresh leaves were gathered from each individual and dried in silica gel at once. According to the phylogeny analyses of Ammopiptanthus by Xie and Yang (2012) and Wang et al. (2006), we selected the species Sophora davidii, S. flavescens, Piptanthus nepalensis, Thermopsis smithiana, T. lanceolata, T. alpina as outgroups for the phylogenic analysis.
DNA extraction, amplification, and sequencing
Total genomic DNA was extracted from dried leaf tissue using a modified 2× CTAB method (Rogers and Bendich 1985; Doyle and Doyle 1987). The ITS region (ITS1, 5.8S rDNA, ITS2) was amplified and sequenced using primers ITS1 and ITS4 according to White et al. (1990), and the spacers trnH–psbA, trnS–trnG, and trnL–trnF were amplified and sequenced using the primers and protocols of Sang et al. (1997), Shaw et al. (2005) and Taberlet et al. (1991) respectively. Amplification products were purified using PCR Product Purification Kits from Shanghai SBS, Biotech Ltd., China. Sequencing reactions were conducted with the forward and reverse primers of the amplification reactions, using the DYEnamic ET Terminator Kit (Amersham Pharmacia Biotech), with an ABIPRISM3730 automatic DNA sequencer from Shanghai Sangon Biological Engineering Technology & Services Co., Ltd (Shanghai, China). Electropherograms were edited and assembled using SEQUENCHER 4.8 (Gene Codes, Ann Arbor, MI, USA). Sequences were aligned using the CLUSTAL W program (Thompson et al. 1994) and refined by visual inspection. The alignments were adjusted manually, and the haplotypes were identified by TCS v.1.21 (Clement et al. 2000).
Phylogenetic divergence time estimation and phylogeographical analysis
Phylogenetic relationships of the cpDNA haplotypes were analysed by maximum parsimony (MP) method implemented in PAUP, version 4.0b10 (Swofford 2002). For MP analyses, the search strategy were as follows: 100 random additions of sequences with tree bisection–reconnection branch swapping, and MULTREES, COLLAPSE, and STEEPEST DESCENT options switched on. We treated indels at positions 542, 1126, 1174, and 2066 as a single mutation event and coded as a substitution. The reliability of the trees was tested using 1000 bootstrapping replicates. Bayesian analysis also used to search for tree topologies and estimated divergence time among the lineages using the software BEAST (Drummond et al. 2002; Drummond and Rambaut 2007). We used the Hasegawa-Kishino-Yano substitution model (selected by MODELTEST) and a constant-size coalescent tree prior. The cpDNA substitution rates for most angiosperm species are uncertain, and have been estimated to vary between 1.0 × 10−9 and 3.0 × 10−9 s s−1 y−1 (Wolfe et al. 1987). We used normal distribution priors with a mean of 2.0 × 10−9 and a SD of 6.080 × 10−10 for Ammopiptanthus to cover this uncertainty within the 95 % range of the distribution for estimation of divergence time of major lineages (Jia et al. 2012). After burning in of 1,000,000 steps, all parameters were sampled once every 1000 steps from 10,000,000 Markov chain Monte Carlo steps. We examined convergence of the stationary distribution by visual inspection of plotted posterior estimates with TRACER (Drummond and Rambaut 2007), and the effective sample size for each parameter was found to exceed 200. Genealogical relationships among all the haplotypes were analysed using statistical parsimony method (Templeton et al. 1992) with a maximum connection limit equal to 20 steps implemented in the program TCS v.1.21 (Clement et al. 2000).
Molecular variability and demographic analysis
HAPLONST was used to estimate within-population diversity (hS), total gene diversity (hT), genetic differentiation (GST) at the species level, and population subdivision for phylogenetically ordered alleles (NST). Spatial analysis of molecular variance was performed to test the spatial genetic structure of cp genetic variation, using SAMOVA v.1.0 (Dupanloup et al. 2002), and the analysis for defining groups of populations (K) was run for K = 2–32. Finally, when the proportion of total genetic variance because of differences among groups of populations (FCT) up to the maxim, the number of groups was accepted as the best grouping pattern of populations. To study the genetic structure, analysis of molecular variance (AMOVA) was employed based on pairwise differences of the sequences, using ARLEQUIN v.3.01 (Excoffier et al. 1992, 2005). To test whether the genetic distance was significantly correlated with geographical distance (Mantel test), pairwise estimates of FST (calculated in ARLEQUIN) and the natural log of geographic distances between locations (calculated in PASSAGE v.1.1; Rosenberg 2001) were correlated using the program IBD v.1.52 (Bohonak 2002).
To test for evidence of range expansions, Tajima’s D and Fu’s FS statistics were calculated in ARLEQUIN (Tajima 1989; Fu 1997). A significant value for D or a significant, large, negative value for FS may represent range expansion (Fu 1997; Aris-Brosou and Excoffier 1996; Tajima 1996). To investigate demographic history, the mismatch distribution (MDA) was also calculated in ARLEQUIN, with 10,000 permutations were performed. An unimodal distribution indicates that populations have experienced a recent expansion (Slatkin and Hudson 1991; Rogers and Harpending 1992). If the sudden expansion model was accepted, we used the relationship s = 2ut to estimate the expansion time (t), where s is the total number of mutations, and u is the mutation rate per generation for the whole analyzed sequence (Rogers and Harpending 1992). The u value was calculated as u = 2 µkg, where µ is the substitution rate per nucleotide site per year (s/s/y), k is the average sequence length of the analyzed DNA region, and g is the generation time in years. We also used the rate 2 × 10−9 for our estimation of range expansion times (Jia et al. 2012). Based on our observation in Turpan Botany Garden, the generation time for this species is 4 years.
Results
Sequence analysis
The aligned sequence length for the trnH–psbA spacer was 340 base pairs (bp), for the trnL–trnF spacer 926 bp, and for the trnS–trnG spacer 918 bp. A total of 36 informative characters were found in the aligned sequence data: 5 nucleotide substitutions (positions 10, 405, 664, 1444, 2004) and 11 indels (positions 196, 197, 314, 323, 542–543, 922, 1126–1130, 1174–1185, 1439, 1561, and 2006–2010). Of the 392 sampled individuals from 33 populations, a total of 8 haplotypes (A–H) were identified (Tables 1, 2). For the ITS region, the aligned sequence length was 687 bp, and 3 informative characters were found in the aligned sequence data, and they are all nucleotide substitutions (positions 40, 443, and 625). Of the 392 sampled individuals from 33 populations, 2 genotypes (a and b) were identified. GenBank accession numbers of the cpDNA sequences are KU178934-KU178946, and of the ITS sequences are KU178932-KU178933.
Genetic variations geographical distribution and relationships
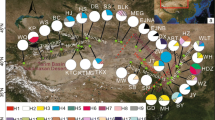
The sampled locations, and geographic distribution of cpDNA haplotypes and ITS genotypes, along with the frequency of them in each population, is presented in Fig. 1 and Table 1. In the network, cp haplotypes A, B, and C are distributed in western Xinjiang (Region 1); haplotypes D–H are distributed in the Alxa Desert (Region 2). No haplotypes were shared between the two regions. In region 1, haplotype A is widespread, and rare haplotypes B and C are fixed in population 5 and 12 respectively. In region 2, haplotype D is widespread on the north of the Alxa Desert, E is fixed in population 22, and haplotypes F, G, and H are restricted to the south (Fig. 1c). For ITS genotype, all populations had one nuclear genotype (a) in region 1, whereas all populations had the other nuclear genotype (b) in region 2 (Fig. 1b).

The network, constructed using parsimony analysis, indicated phylogeny among the haplotype. As shown in Fig. 2, haplotype B was connected to A by one hypothetical haplotypes, and haplotype C was connected to A by one substitution. Haplotype A was connected F by 9 hypothetical haplotypes, and F was connected to D by one hypothetical haplotypes. Haplotype G was connected to H by one substitution, which in turn was connected to D by one hypothetical haplotypes. Haplotype E was connected to D by one substitution.

Cp haplotype network of Ammopiptanthus constructed under the criterion of statistical parsimony. In the haplotype parsimony network, the blank circles indicate the missing or inferred haplotypes; the circle size is proportional to haplotype frequencies; haplotypes in the network showed in the same colours correspond to those in the geographical distribution, Fig. 1c
Genetic diversity and genetic structure
Spatial genetic analysis of cpDNA haplotypes using SAMOVA indicated that FCT increased to a maximal value of 0.9842 when K (the number of groups) was raised from K = 2 to K = 4. The grouping pattern of populations corresponding to K = 4 is: (1) populations 1–12, belonging to western Xinjiang; (2) populations 13–26, 28–29, belonging to Inner Mongolia and Ningxia province; (3) population 27, belonging to Inner Mongolia; (4) populations 30-33, belonging to Inner Mongolia, Ningxia, and Gansu province. For the whole distribution of Ammopiptanthus, within-population gene diversity (hS) was 0.045 (SE 0.0221), and total gene diversity (hT) was 0.670 (SE 0.0522). Differentiation among populations was high (GST = 0.932, SE 0.0315), indicating significant population structure. NST was 0.981 (SE 0.0148), significantly higher than GST as shown by the U test (U = 1.96, p < 0.001), indicating significant phylogeographic structure. In the Alxa Desert, hS was 0.057 (SE 0.0318), hT was 0.434 (SE 0.1248), and differentiation among populations was high (GST = 0.869; NST = 0.904). In western Xinjiang, hS was 0.039 (SE 0.0277), hT was 0.041 (SE 0.0289), and differentiation among populations was low (GST = 0.056; NST = 0.073). The AMOVA results provide evidence that 98.00 % (p < 0.001) of the total variation can be explained by differences among populations. When populations were grouped according to genetic difference and geographical region by the program SAMOVA, AMOVA results demonstrated that 98.44 % (p < 0.001) of the total variation occurred among the groups (Table 3). According to geographic regions, 89.81 % (p < 0.001) of the total variation occurred between the two regions. Mantel’s test showed that there was significant correlation between genetic distance and geographical distance (r = 0.757, p < 0.0001, Fig. 3).

The figure shows a significant relationship between geographic and genetic distance (r = 0.757, p < 0.0001)
Demography of the groups in Ammopiptanthus
Demographic analysis of groups and total individuals in Ammopiptanthus revealed that, group 1 (populations 1–12 in western Xinjiang) experienced range expansion in the past. Range expansion of group 1 is supported by significant results of Fu’s FS (Fs = −2.83, P = 0.006), along with unimodal distributions for the shapes of the mismatch distribution (Fig. 4). The time of the geographic range expansion of group 1 is estimated to have occurred at about 4.29 × 104 years, which is consistent with the Interstadial of the Last glaciation (Shi et al. 2005).

Mismatch distribution analysis for chloroplast DNA data for group 1 (Ammopiptanthus nanus) that includes populations 1–12 in western Xinjiang (SSD = 0.001, p = 0.04)
Phylogenetic analysis and divergence time between major clades
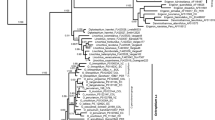
MP and Bayesian analysis produced trees with the same topology, and so only the MP tree is presented (Fig. 5). In this tree, Ammopiptanthus is resolved as monophyletic, and the relationship is well supported (100 % bootstrap support and 1.00 posterior probability). The two intraspecific lineages of Ammopiptanthus were estimated to have diverged between 0.77 (95 % HPD 0.35–1.35) Mya, that is consistent with the Maximum Glaciation in early-middle Pleistocene, according to Shi et al. (2005).

Phylogenetic relationships of 8 haplotypes of Ammopiptanthus and related species. Numbers above branches are support values, the front is maximum parsimony bootstrap values greater than 50, and the latter is posterior probabilities greater than 0.9. The estimated divergence time is approximately 0.77 Mya at node 1
Discussion
Allopatric divergence between two regions of Ammopiptanthus in northwestern China
A single lineage of ancestral descendant populations of organisms which maintains its identity from other such lineages and which has its own evolutionary tendencies and historical fates, should be defined as species status (Wiley 1978). For both the nuclear and chloroplast datasets, phylogenetic analyses showed that Ammopiptanthus comprised two lineages, distributed respectively in western Xinjiang (Region 1) and the Alxa Desert (Region 2) (Figs. 1b, c, 2). Parsimony analysis also indicated the similar result (Fig. 2). The two reciprocally monophyletic clades in Ammopiptanthus give further molecular support to the suggestion that populations in western Xinjiang should be given species rank, A. nunas (Cui 1998).
AMOVA analysis and the Mantel test demonstrated significant genetic divergence between the two regions. Based on molecular dating analysis, Xie and Yang (2012) confirmed the previous hypothesis that Ammopiptanthus in eastern central Asian deserts is an early Miocene relic genus, whose ancestor was probably one of the elements of the xerophytes in the Tethyan flora (Liu 1995; Sun 2002; Sun and Li 2003), thus, we infer that the species diffused into the Alxa desert through the conduit Hexi Corridor during the Tertiary period (Liu 1995). About 2 Mya, the temperature declined markedly in northwest China when the Quarternary came (Guo et al. 1999). Despite of the developing cold climate, it appears that Ammopiptanthus could have survived in some valleys of the western Tianshan Mountains in Xinjiang. Likewise, the Alxa Desert resides greatly inland and was not covered by glaciers, thus it suffered relatively little damage from the cold climate (Liu 1995), and is therefore another habitat in which this species would have survived during the climatic extremes. Aridification usually correlated with periodicly cold glacial episodes (Shi et al. 2005; Xu et al. 2010). From the early to middle Pleistocene, dry climate continued to develop, and deserts and gobi deserts further expanded in northwest China (Williams et al. 1993). At 0.8–0.6 Mya, north-western China entered into the Maximum Glaciation, the climate became much colder and dryer beyond the extent of former periods (Williams et al. 1993; Shi et al. 2005). The lineages of Ammopiptanthus (node 1 in Fig. 5) diverged approximately 0.77 Mya, consistent with this period, thus, we speculate that the extreme climate reduced and isolated acceptable habitats for the species. In addition, between the two geographic regions, there are the vast Taklimakan desert and long Hexi Corridor, with a distance exceeded 2700 km, interrupting the gene flow and leading to the allopatric divergence. Allopatric divergence correlated with extremely low temperatures during glacial periods was also found in plants on the QTP, such as Hippophae tibetana and Aconitum gymnandrum, with major lineages of them all split for the coldness in the early Pleistocene (Wang et al. 2009; Jia et al. 2011).
Genetic diversity and genetic structure in the two species of Ammopiptanthus
The level of genetic diversity in the two species are both low, consistent with the previous study in Ammopiptanthus, using different molecular markers (ISSR, AFLP, and allozyme) (Ge et al. 2005; Chen et al. 2009a, b). The association between low genetic variation and endemism and/or rarity has long been known (Frankham et al. 2002). Several factors might account for the low level of genetic diversity. First, A. mongolicus and A. nanus may originate from genetically depauperate populations, the low inherent variability of the ancestral species might be the basic reason for the low level of genetic diversity (Ge et al. 2005). Second, small and isolated populations of the two species were apt to suffer from genetic drift, that may have contributed to the lack of genetic diversity observed today (Ge et al. 2005). Finally, selfing were both found in the two species (Chen et al. 2009a, b). They have numerous flowers on a single inflorescence, about 10–16 flowers per inflorescence. Large plants with many flowers can induce the pollinator to visit many flowers per plant, thereby increasing the probability of selfing, either by pollinator-mediated self-pollination or geitonogamy (Ge et al. 2005; Chen et al. 2009a; Burdon et al. 1988; Wolff et al. 1988).
According to Gitzendanner and Soltis (2000), in widespread and restricted congeners, genetic variation was significantly lower in the rare species than in the widespread species. Compared with Ammopiptanthus mongolicus, lower genetic diversity and structure in A. nanus could be related to its more limited geographical ranges. For A. mongolicus, haplotype D is widespread in the north of the Alxa Desert, apparently different from the genetic variation in the south of the desert. The present genetic distribution in the north of the desert could be a result of the rapid expansion of the species from a nearby refugium (Chen et al. 2009a). Origins from different refugia may be responsible for the genetic divergence between north and south of the Alxa Desert, which was consistent with the results based on allozyme by Chen et al. (2009a).
Regional expansion in western Xinjiang
Range expansion of western Xinjiang (group 1) is supported by significantly negative Fu’s FS value, and unimodal mismatch distribution (Fig. 4). Whenever large-scale range expansion occurs, it usually causes two distinct genetic signatures: wide distribution of a single genotype, and a star-phylogeny pattern (a dominant haplotype with multiple rare alleles) (Comes and Kadereit 1998; Hewitt 2000). The distribution of genetic variation in western Xinjiang was consistent with these expected signatures (Figs. 1, 2). We found that haplotype A was widespread across every sampled location of the region/group, and rare haplotypes B and C were limited to random single populations of the valleys in the western Tianshan Mountains.
Climate oscillation since the late Quaternary is usually considered an important factor influencing the current geographical distribution and genetic structure of plants (Hewitt 2004). Fluctuant climate can cause corresponding range shifts. There was a great deal of glaciation developing in the high Tianshan Mountains during the glaciation, and the extent of glacial area varied in response to the alternation of glaciation and interglaciation (Shi et al. 2005). As noted, the time of expansion in western Xinjiang is consistent with the Interstadial of the Last glaciation (Shi et al. 2005). During the interglacial period, environmental conditions became warmer and, with the increasing temperature, a greater amount of run-off from melting glacial ice in the high mountains infused the valleys, and thus made habitats there more moist (Williams et al. 1993). The improved habitats are more suitable for recovery, then the species would have thrived and expanded outwards. There was a hypothesis proposed in published studies, that was A. nanus might originate from a founder population containing only a small fraction of the genetic variation present in its progenitor species, and it might experienced founder events or bottleneck effect in its evolutionary history (Ge et al. 2005; Chen et al. 2009b). Our study confirmed the hypothesis, the variant haplotype A might surf on the wave of advance of the refugium range expansion, reaching high frequencies, spreading to the other suitable habitats in valleys of the western Tianshan Mountains.
Implications of conservation in Ammopiptanthus
Genetic drift and inbreeding are the most important genetic factors leading to a loss of genetic diversity in species with small population sizes and fragmented distribution patterns and pushing them into “extinction vortices” (Lande 1998). The maintenance of genetic diversity is a critical issue in the conservation for long-term survival and maintain evolutionary potential of threatened species (Frankel 1983). According to the results of this study, efforts for recovery should be implemented at the scale of populations in the two species. First, we should adopt in situ conservation for the two species. Although several nature reserves, such as the west Ordos, the Helan Mountain, the Daqing Mountains, and the Urat nature reserves have been set up for A. mongolicus in Inner Mongolia, such nature reserves should likewise be set up in Ulugqat Country in Xinjiang as well. Second, actions such as transplantation of seedlings in natural habitats and/or ex situ conservation should be taken in order to create augmented space for genetic exchange and recombination. In addition, populations harbour relatively high amounts of the genetic diversity or rare genetic variations in each species should be prior considered, such as populations Biaoertuokeyi and Atushi in A. nanus, populations Wulanbuhe, Beisi, Lingwu, Jiergelang, and Jingtai in A. mongolicus.
References
Aris-Brosou S, Excoffier L (1996) The impact of population expansion and mutation rate heterogeneity on DNA sequence polymorphism. Mol Biol Evol 13:494–504
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge
Birky CW Jr (2001) The inheritance of genes in mitochondrial and chloroplast: laws, mechanisms, and models. Annu Rev Genet 35:125–148
Bohonak AJ (2002) IBD (isolation by distance): a program for analyses of isolation by distance. J Hered 93:153–154
Browning TL, Taggart DA, Rummery C, Close RL, Eldridge MDB (2001) Multifaceted genetic analysis of the ‘‘Critically Endangered’’ brush-tailed rock-wallaby Petrogale penicillata in Victoria, Australia: implications for management. Conserv Genet 2:145–156
Burdon JJ, Jarosz AM, Brown AHD (1988) Temporal patterns of reproduction and outcrossing in weedy populations of Echium plantagium. Biol J Linn Soc 34:81–92
Chen GQ, Crawford D, Huang HW, Ge XJ (2009a) Genetic structure and mating system of Ammopiptanthus mongolicus (Leguminosae), an endangered shrub in north-western China. Plant Species Biol 24:179–188
Chen GQ, Huang HW, Crawford DJ, Pan BR, Ge XJ (2009b) Mating system and genetic diversity of a rare desert legume Ammopiptanthus nanus (Leguminosae). J Syst Evol 47(1):57–66
Clement M, Posada D, Crandall KA (2000) TCS: a computer program to estimate gene genealogies. Mol Ecol 9:1657–1659
Comes HP, Kadereit JW (1998) The effect of quaternary climatic changes on plant distribution and evolution. Trends Plant Sci 3:432–438
Crandall KA, Bininda-Emonds ORP, Mace GM, Wayne RK (2000) Considering evolutionary processes in conservation biology. Trends Ecol Evol 15:290–295
Cui HB (1998) Fabaceae (5). In: Wu ZY, Raven PH (eds) Flora of China, vol 42. Science Press, Beijing, pp 395–397
Dang RL, Pan XL, Gu XF (2002) Floristic analysis of spermatophyte genera in the arid deserts area in North-West China. Guihaia 22(2):121–128
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure from small quantities of fresh leaf tissues. Phytochem Bull 19:11–15
Drummond AJ, Rambaut A (2007) BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214
Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W (2002) Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161:1307–1320
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define the genetic structure of populations. Mol Ecol 11:2571–2581
Excoffier L, Smouse P, Quattro J (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: applications to human mitochondrial DNA restriction data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Frankel OH (1983) The place of management in conservation. In: Schonewald-Cox CM, Chambers SM, MacBryde B, Thomas WL (eds) Genetics and conservation. Benjamin-Cummings, Menlo Park
Frankham R, Ballou JD, Briscoe DA (2002) Introduction to conservation genetics. Cambridge University Press, Cambridge
Fraser DJ, Bernatchez L (2001) Adaptive evolutionary conservation: towards a unified concept for defining conservation units. Mol Ecol 10:2741–2752
Fu LG (1992) Rare and endangered plants in China. Science Press, Beijing, pp 368–370
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking, and background selection. Genetics 147:915–925
Ge XJ, Yu Y, Yuan YM, Huang HW, Yan C (2005) Genetic diversity and geographic differentiation in endangered Ammopiptanthus (Leguminosae) populations in desert regions of northwest China as revealed by ISSR analysis. Ann Bot (London) 95:843–851
Gitzendanner MA, Soltis PS (2000) Patterns of genetic variation in rare and widespread plant congeners. Am J Bot 87:783–792
Guo ZT, Peng SZ, Hao QZ, Chen XH, Liu TS (1999) Late Tertiary development of aridification in northwestern China: link with the arctic ice-sheet formation and Tibetan uplifts. Quat Sci 6:556–566 (in Chinese with English abstract)
Guo YP, Zhang R, Chen CY, Zhou DW, Liu JQ (2010) Allopatric divergence and regional range expansion of Juniperus sabina in China. J Syst Evol 48:153–160
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hewitt GM (2004) Genetic consequences of climatic oscillations in the Quaternary. Phil Trans R Soc Lond B Biol Sci 359:183–195
Jia DR, Liu TL, Wang LY, Zhou DW, Liu JQ (2011) Evolutionary history of an alpine shrub Hippophae tibetana (Elaeagnaceae): allopatric divergence and regional expansion. Biol J Linn Soc 102:37–50
Jia DR, Abbott RJ, Liu TL, Mao KS, Bartish IV, Liu JQ (2012) Out of the QinghaieTibet Plateau: evidence for the origin and dispersal of Eurasian temperate plants from a phylogeographic study of Hippophaë rhamnoides (Elaeagnaceae). New Phytol 194:1123–1133
Lande R (1998) Anthropogenic, ecological and genetic factors in extinction and conservation. Res Popul Ecol 40:259–269
Li ZH, Chen J, Zhao GF, Guo YP, Kou YX, Ma YZ, Wang G, Ma XF (2012) Response of a desert shrub to past geological and climatic change: a phylogeographic study of Reaumuria soongarica (Tamaricaceae) in western China. J Syst Evol 50:351–361
Liu YX (1995) A study on origin and formation of the Chinese desert floras. Acta Phytotaxon Sin 33:131–143
Liu JQ, Sun YS, Ge XJ, Gao LM, Qiu YX (2012) Phylogeographic studies of plants in China: advances in the past and directions in the future. J Syst Evol 50:267–275
Mäder G, Zamberlan PM, Fagundes NJR, Magnus T, Salzano FM, Bonatto SL, Freitas LB (2010) The use and limits of ITS data in the analysis of intraspecific variation in Passiflora L. (Passifloraceae). Genet Mol Biol 33:99–108
Martins K, Chaves L, Vencovsky R, Kageyama P (2011) Genetic structure based on nuclear and chloroplast microsatellite loci of Solanum lycocarpum A. St. Hil. (Solanaceae) in Central Brazil. Genet Mol Res 10:665–677
Moritz C (1994) Applications of mitochondrial DNA analysis in conservation: a critical review. Mol Ecol 3:401–411
Osborne MJ, Norman JA, Christidis L, Murray ND (2000) Genetic distinctness of isolated populations of an endangered marsupial, the mountain pygmy-possum, Burramys parvus. Mol Ecol 9:609–613
Qin AL, Wang MM, Cun YZ, Yang FS, Wang SS, Ran JH, Wang XQ (2013) Phylogeographic evidence for a link of species divergence of Ephedra in the Qinghai-Tibetan Plateau and adjacent regions to the Miocene Asian Aridification. PLoS One 8(2):e56243
Rogers SO, Bendich AJ (1985) Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant-tissues. Plant Mol Biol Rep 5:69–76
Rogers A, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Rosenberg MS (2001) PASSAGE: Pattern Analysis, Spatial Statistics and Geographic Exegesis. Ver. 1.1. Department of Biology: Arizona State University
Ryder OA (1986) Species conservation and systematics: the dilemma of subspecies. Trends Ecol Evol 1:9–10
Sang T, Crawford DJ, Stuessy TF (1997) Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am J Bot 84:1120–1136
Shaw J, Lickey E, Beck JT, Farmer SB, Liu W, Miller J, Siripun KC, Winder CT, Schilling EE, Small RL (2005) The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am J Bot 92:142–166
Shi YF, Cui ZJ, Su Z (2005) The quaternary glaciations and environmental variations in China. Hebei Science and Technology Publishing House, Hebei, China, pp 85–100
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Su ZH, Zhang ML (2013) Evolutionary response to Quaternary climate aridification and oscillations in northwestern China revealed by chloroplast phylogeography of Nitraria sphaerocarpa (Nitrariaceae). Biol J Linn Soc 109:757–770
Su ZH, Zhang ML, Sanderson SC (2011) Chloroplast phylogeography of Helianthemum songaricum (Cistaceae) from northwestern China: implications for preservation of genetic diversity. Conserv Genet 12:1525–1537
Sun H (2002) Tethys retreat and Himalayas-Hengduanshan Mountains uplift and their significance on the origin and development of the Sino-Himalayan elements and alpine flora. Acta Bot Yunnanica 24:273–288
Sun H, Li ZM (2003) Qinghai-Tibet plateau uplift and its impact on Tethys flora. Adv Earth Sci 18:852–862
Swofford DL (2002) PAUP*: phylogenetic analysis using parsimony (and other methods), Version 4.0b10. Sinauer Associates, Sunderland, MA
Taberlet PL, Pautou GG, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tajima F (1996) The amount of DNA polymorphism maintained in a finite population when the neutral mutation rate varies among sites. Genetics 143:1457–1465
Tang YC (2000) On the affinities and the role of the Chinese flora. Acta Bot Yunnanica 22:1–26
Templeton AR, Crandall KA, Sing CF (1992) A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 132:619–633
Thompson JD, Higgins DG, Gibson TJ (1994) Clustal-W—improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Wang HC, Sun H, Compton JA, Yang JB (2006) A phylogeny of Thermopsideae (Leguminosae: Papilionoideae) inferred from nuclear ribosomal internal transcribed spacer (ITS) sequences. Biol J Linn Soc 151:365–373
Wang LY, Abbott RJ, Zhang W, Chen P, Wang YJ, Liu JQ (2009) History and evolution of alpine plants endemic to the Qinghai-Tibetan Plateau: Aconitum gymnandrum (Ranunculaceae). Mol Ecol 18:709–721
Wei Z, Lock JM (2010) Fabaceae Tribe Thermopsideae. In: Wu ZY, Raven PH (eds) Flora of China, vol 10. Science Press, Beijing, p 100
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic Press, San Diego, pp 315–322
Wiley EO (1978) The evolutionary species concept reconsidered. Syst Biol 27:17–26
Williams MAJ, Dunkerley DL, De Dekker P, Kershaw AP, Stokes T (1993) Quaternary environments. Edward Arnold, London
Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAS. Proc Natl Acad Sci USA 84:9054–9058
Wolff K, Frisco B, van Damme JMM (1988) Outcrossing rates and male sterility in natural populations of Plantago coronopus. Theor Appl Genet 76:190–196
Xie L, Yang Y (2012) Miocene origin of the characteristic broad-leaved evergreen shrub Ammopiptanthus (Leguminosae) in the desert flora of eastern central Asia. Int J Plant Sci 173(8):944–955
Xu X, Kleidon A, Miller L, Wang SQ, Wang LQ, Dong GC (2010) Late Quaternary glaciation in the Tianshan and implications for palaeoclimatic change: a review. Boreas 39:215–232
Acknowledgments
This research was supported by the National Natural Science Foundation of China (No. 41271070) and Xinjiang Institute of Ecology and Geography, Chinese Academy of Sciences. We thank professor Rui Cao and professor Yizhi Zhao at college of life science in Inner Mongolia University for their kind help in sampling work in Inner Mongolia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Su, Z., Pan, B., Zhang, M. et al. Conservation genetics and geographic patterns of genetic variation of endangered shrub Ammopiptanthus (Fabaceae) in northwestern China. Conserv Genet 17, 485–496 (2016). https://doi.org/10.1007/s10592-015-0798-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-015-0798-x