
Abstract
The great scallop Pecten maximus is harvested in several European countries and fisheries targeting this species are severely regulated by fishing quotas. In addition, hatchery-based population enhancement has been developed in some countries to provide alternative or complementary production. The genetic structure of wild populations of P. maximus and the potential impact of aquaculture on the genetic diversity of this species remains poorly documented. In this study, we explored the genetic structure of P. maximus using 12 microsatellite markers, considering 14 populations sampled from Galicia (Spain) to the North of Norway, and one population of Pecten jacobaeus (L., 1758) from the Lion Gulf (Mediterranean Sea). Results indicated a clear differentiation between Norwegian and Atlantic (from Ireland to Spain) populations, but very little to no difference between populations within these two groups. A decrease of the genetic diversity was observed with latitude. No significant reduction of the genetic diversity was observed in the Bay of Brest, where hatchery-based population enhancement has been performed intensively since 1983. Our results are discussed in the light of the inferred recent evolutionary history, phylogeography and connectivity of populations in Europe, and of the phenotypic variability reported in previous studies between northern and southern populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The great scallop, Pecten maximus (L., 1758), is a benthic marine bivalve, which is broadly distributed in shallow waters along the European coasts, from northern Norway to southern Spain. This species is of high economical value, mainly in U.K. and France, with landings reaching 63,681 tonnes in 2012 (Beaumont and Gjedrem 2006; FAO 2015). Although dredging of natural beds remains the main method of exploitation, sea-ranching has emerged as an alternative or complementary production solution. This is notably the case in the Bay of Brest (Brittany, France), where sea-ranching of P. maximus has been carried out since 1983. Following a particularly cold winter in 1962–1963 (Dao et al. 1999), stocks were severely depleted and sea-ranching was ultimately proposed to revive the local fishery (Dao et al. 1985). Hatchery production of juvenile great scallops is now well mastered, although several aspects could still be optimized (Robert and Gérard 1999; Andersen et al. 2011), and a few commercial hatcheries market significant quantities of scallop seed: 1–5 million seeds are produced yearly by Le Tinduff hatchery (Plougastel, France). Similar initiatives are in development in northern Europe.
It is known that hatchery practices can significantly impact the effective population size of enhanced stocks, especially in bivalve species for which variance in reproductive success has been shown to be large (e.g. Boudry et al. 2002). Restocking has been historically low in P. maximus, and is now linked to spatially limited aquaculture practices (Beaumont 2000; Minchin 2003). Thus, the impact of transplantations and hatchery stocks on natural populations is therefore presumed to be low (Gaffney 2006; Hold et al. 2012). However, this issue has not been addressed thoroughly, yet. In France, the Bay of Brest is of special interest in terms of conservation, as population enhancement based on seed produced in the local hatchery (Le Tinduff, Plougastel, France) is well developed, and might have a significant impact on the local genetic diversity (Beaumont and Gjedrem 2006). Indeed, landings of hatchery-born scallops are estimated to be around 2/3 of total landings in the Bay of Brest (Fleury et al. 2005).
Knowledge of the genetic structure of wild populations is a key step to quantify the potential impact of restocking and aquaculture practices on natural resources. Genetic differentiation patterns over the whole geographic distribution of populations are shaped by multiple forces during the evolutionary history of species (e.g. gene flow, genetic drift, selection). In addition to natural forces, the genetic variability of wild populations can be affected by anthropogenic pressures (e.g. pollution, habitat fragmentation, translocations, harvesting) (Smith et al. 1991; Ma et al. 2000). The great scallop, like many bivalve mollusks, can disperse over broad distances during its long planktonic larval phase (Le Pennec et al. 2003). In the open sea, estimates of the planktonic larval duration (PLD) are 18–42 days (Le Pennec et al. 2003). PLD is strongly influenced by water temperature, and larvae take a longer time to achieve metamorphosis in cold water. Hence, in laboratory condition, PLD has been assessed around 24–35 days at 18 °C, and up to 65–78 days at 8–9 °C (Beaumont and Barnes 1992). Thus, larvae clearly display a high dispersal potential, which may be further increased in colder waters. Additionally, newly metamorphosed post-larvae can also migrate via byssus-drifting (Beaumont and Barnes 1992). A larval dispersal model developed by Nicolle et al. (2013) has revealed an important dispersal potential at a small geographic scale. With regards to these results, population genetics could provide insights into gene flow patterns along the latitudinal gradient: gene flow might be higher in the northern than in the southern range of the species. Limited data are available about the population genetic structure of P. maximus along its distribution area. Genetic structuring has been described between Norway and the British Isles, using mitochondrial DNA (Ridgway and Dahle 2000). Local studies have also been conducted, revealing a rather consistent pattern of population structuring with a large general homogeneity along Atlantic coasts, using allozymes (Beaumont et al. 1993) or mitochondrial DNA (Heipel et al. 1998, 1999; Wilding et al. 1997). Some hints about small scale structuring have been found with mitochondrial DNA in specific locations, i.e. Mulroy Bay (Wilding et al. 1997; Heipel et al. 1998, 1999) and east of the Isle of Man (Heipel et al. 1998, 1999).
Further studies have recently been conducted on phenotypic traits, suggesting a significant differentiation among great scallop populations. Chauvaud et al. (2012) have described differentiated growth patterns between “northern” and “southern” populations, with individuals from northern populations showing a slower growth but reaching a larger asymptotic size and a lower number of growth days per year. The authors suggested a possible effect of phenotypic plasticity without excluding a possible local adaptation associated with a genetic differentiation of populations. Another type of phenotypic variation, related to the timing of reproduction, has been observed between populations from Scotland, the Bay of Brest and the Bay of Saint Brieuc (France). Features of the reproductive cycle of transplanted scallops remained unchanged following population translocations, suggesting a genetic basis to these traits (Paulet et al. 1988; Cochard and Devauchelle 1993; Mackie and Ansell 1993). A similar maintenance of the natural reproductive cycle has been reported in Norway with scallops transferred between populations along the coast (Magnesen and Christophersen 2008). More recently, a proteomic study has revealed phenotypic differences between two populations sampled in Norway and France (Artigaud et al. 2014). However, the genetic architecture of these phenotypic differences remains to be studied.
A sister species of P. maximus has been described in the Mediterranean Sea, Pecten jacobaeus (L. 1758). Several genetic studies have suggested that their phylogenetic status is unclear, showing low genetic (allozymes, 16S mtDNA and mitochondrial RFLPs) and phenotypic (shell morphology) differentiation between the two taxa (Wilding et al. 1999; Saavedra and Peña 2004, 2005), leading authors to conclude that they could be considered as a single species. In this context, the relative status of these two taxa remains to be further characterized using microsatellites.
The present study reports a large scale population genetics study of P. maximus, based on 12 microsatellite markers. Populations from Norway to Spain, including the Bay of Brest, are considered in the analysis, as well as a Mediterranean population. This study is motivated by two objectives: (1) assessing the genetic structure of P. maximus along the European coasts and, and (2) giving a first insight of the influence of restocking practices on this structure.
Materials and methods
Studied populations
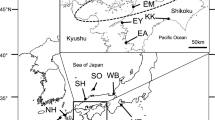
Fifteen populations were sampled from Bodø (Grønholmen, Norway) to the Gulf of Lion (France) (Fig. 1). Twenty-four to forty-eight individuals were collected in each population sample (see supplementary material for details). A fragment of adductor muscle was sampled from each scallop and either preserved in 85 % ethanol or frozen at −20 °C.

Sampling map. Population sampling location from south to north: MPJ Mediterranean Pecten jacobaeus Lion Gulf, France; RDA Ría de Arousa, Galicia, Spain.; RLB Ronce-Les-Bains, La Tremblade, France; BQB Bay of Quiberon, France; RDB Bay of Brest, France. BSB Bay of Saint Brieuc, France; PEB Port-en-Bessin, France; PLY Plymouth, England; IRL Irish Sea: NRV Rennesøy, Vignesholmane, Norway; NTH Træna, Husøya, Norway; NBH Brønnøysund, Hensteinen, Norway; NVR Vikna, Ravsholmen, Norway; NFB Froan, Bordsholmen, Norway; NBG Bodø, Grønholmen, Norway
In the Bay of Brest, where hatchery-raised scallops are released in the wild, the natural origin of individuals was confirmed by checking the absence of a double growth ring on the shells, characteristic of hatchery-born individuals. A double growth ring results from a period of slow growth due to the stress associated with the drastic environmental change during the transfer from the hatchery cage to the natural sea bed (Fleury et al. 2005).
DNA extraction and microsatellite genotyping
DNA extraction was performed using the QIAamp DNA Mini Kit (Qiagen™, Hilden, Germany), according to manufacturer’s instructions. DNA concentrations were estimated using Nanodrop®, and then diluted to 10 ng DNA/mL.
Genetic variation at 12 microsatellite loci was assayed, including two multiplex sets of four markers each from Morvezen et al. (2013) (mx1 and mx2) and one newly developed multiplex of four additional microsatellites named mx4 (see supplementary material for details). The latter was developed following the procedure described in Morvezen et al. (2013) (see supplementary materials for loci statistics and multiplex parameters). Multiplex PCRs were conducted as described in Morvezen et al. (2013).
Briefly, PCR amplifications were conducted with the Type-it Microsatellite PCR Kit (Qiagen TM) in a 10 µl reaction volume containing 5 µl of Type-it Multiplex PCR Master Mix 2X (including HotStarTaq © Plus DNA Polymerase, Type-it Microsatellite PCR Buffer with MgCl2, and dNTPs), 1 µl primer mix (see Morvezen et al. 2013 and supplementary materials for details), 1 µl Q-solution 5X, 2 µl RNase-free water and 1 µl diluted genomic DNA (10 ng). PCRs included an initial step: 95 °C for 15 min, (94 °C 30 s, 59 °C 90 s and 72 °C 90 s) × 30 cycles, followed by a “nested” step (94 °C 30 s, 55 °C 90 s and 72 °C 90 s) × 8 cycles.
PCR products were mixed with Hi-Di formamide and GeneScan 500-LIZ size standard (Applied Biosystems™ Carlsbad, CA, USA): 1 µl PCR product, 10 µl Hi-Di Formamide, 0.15 µl GS500-LIZ. After five minutes of denaturation at 96 °C and a rapid cooling on ice, PCR products were electrophoresed on an ABI-3130 capillary sequencer (Applied Biosystem™). Fragment lengths and allele scoring were assessed with Genemapper 4.0 software (Life Technologies™).
Data analysis
Within-population diversity
Allelic richness, observed and expected heterozygosities, and locus specific F IS were estimated using Fstat 2.9.3.2 (Goudet 1995). Population specific F IS were calculated using Genetix 4.05 (Belkhir et al. 2001). The significance of F IS estimates was tested using 10,000 permutations, and a false discovery rate (FDR) correction for multiple testing was applied. The presence of null alleles was estimated using Microchecker 2.2.3 (Van Oosterhout et al. 2004). Multi-locus allelic richness, observed and expected heterozygosities were plotted against latitude to explore a possible latitudinal gradient in the genetic diversity, and correlation was tested using Pearson’s correlation test with R 2.15 (R Core Team 2012). Linkage disequilibrium for each pair of marker in each population was estimated using Genepop 4.0.5 (Rousset 2008). Effective population sizes were estimated using the linkage disequilibrium method implemented in LDNe 1.31 (Waples and Do 2010); P crit was set at 0.02, as recommended by the authors.
Population genetic structure
Global and pairwise F ST were estimated using Genetix 4.05, and significance was tested using 10,000 permutations. P values were adjusted using FDR correction for multiple testing. A multidimensional scaling (MDS) analysis was computed in R 2.15, using F ST (Weir and Cockerham 1984) linearized according to Slatkin’s transformation (Slatkin 1995). Slatkin’s transformation (FST/(1−FST)) allow FST to behave more like an Euclidian distance and thus is better suited to do MDS. Exact tests of genetic differentiation were conducted with Genepop 4.0.5 with 1000 dememorisations, 100 batches and 10,000 iterations per batch. Bayesian estimations of F ST were computed with EBFST v1.0 (Kitada et al. 2007).
An Analysis of Molecular Variance (AMOVA) was performed with Arlequin 3.5 (Excoffier and Lischer 2010). Populations were aggregated into two clusters, found with the MDS analysis. The first contained Atlantic populations (RDA, RLB, BQB, RDB, BSB, PEB, PLY, IRL), and the second contained Norwegian populations (NRV, NFB, NVR, NBH, NTH, NBG); the Mediterranean population was excluded from this analysis. The genetic structure was assessed by variance analysis within individuals (F it ), among groups (F ct ), among populations within groups (F sc ) and among individuals within populations (F IS) and the significance of the fixation indices was tested with 10,000 permutations. Allelic richness, heterozygosities and F IS were also compared between those two clusters with Fstat 2.9.3.2.
A Mantel test was performed to test isolation by distance (IBD) with Genetix 4.05 using the same genetic distance matrix as used in the MDS, and a matrix of geographic distances among populations. Geographic distances were assessed following the coast line, or, when not possible, following the shortest marine distance. The Mantel test was performed on all populations (excluding the Mediterranean population because of its outlier status, both genetically and geographically) and within each cluster of populations defined above (i.e. Atlantic and Norwegian populations), and significance was assessed by 10,000 permutations.
Results
Genetic diversity within populations
Multilocus allelic richness (A r ) ranged from 6.71 (NBH) to 8.43 (MPJ) (Fig. 2). Observed multilocus heterozygosity (H o ) varied from 0.60 (NBG, NTH, NBH) to 0.67 (MPJ & RDB). Although all markers were initially developed for Atlantic scallops, the highest mean A r was observed in the Mediterranean population (A r = 8.43) and the lowest was found in the NBG, NTH and NBH samples (A r = 6.71–6.97; see supplementary material for details). Among P. maximus samples, there was a significant decrease in A r with increasing latitude, as illustrated in Fig. 2 (Pearson correlation test, r = −0.92, p < 0.001). A similar trend was observed for H o (data not shown, r = −0.75, p = 0.002). This latitudinal decrease in genetic diversity was not observed within either the Atlantic and Norwegian groups of populations (as defined below). No significant pattern of linkage disequilibrium was found: less than 5 % of each pair of marker tested on each population gave a significant result.

Allelic Richness plotted against latitude. Site acronym signification is given in Fig. 1
A moderate but significant heterozygote deficiency was found over all loci (see supplementary material). Five loci in particular showed recurrent heterozygote deficiencies across most populations (after FDR correction for multiple testing): PmRM007 and PmRM027 (five significant F IS estimates), PmRM71 (six significant F IS estimates), PmRM012 (eight significant F IS estimates) and PmRM043 (ten significant F IS estimates) (see supplementary material for details). The presence of null alleles was detected by Microchecker at three loci: PmRM007, PmRM027 and PmRM043. The estimated frequencies of null alleles for these three loci were respectively 0.06, 0.14 and 0.19. Interestingly, no null alleles were detected for PmRM012 and PmRM71.
Confidence intervals of all Ne estimates included infinity, which did not allow us to have a clear estimate of effective population size. However, the method used should not have any difficulties in estimating Ne if it was small (Waples and Do 2010); thus, we can at least conclude that Ne was large to very large in all our populations.
Population genetic structure
The Weir and Cockerham (1984) and the bayesian (Kitada et al. 2007) estimations of F ST gave similar results (data not shown). Only Weir and Cockerham (1984) F ST are described and discussed hereafter.
The global level of differentiation among all population samples was relatively high (F ST = 0.0291, p < 0.001). We then tested if null alleles had a significant influence on the genetic structure by dropping the three loci with a high proportion of null alleles. Global F ST and confidence intervals were similar (Respectively, global F ST = 0.0291 (CI 95 % 0.0157–0.0472) and F ST = 0.0287 (CI 95 % 0.0160–0.0424), so all further results are given for all loci. All pairwise F ST comparisons including the Mediterranean population (MPJ) were significant (0.03 < F ST < 0.08) and generally higher than estimates among P. maximus populations (−0.05 < F ST < 0.06) (Table 1). Moreover, all pairwise F ST comparisons between Norwegian and Atlantic P. maximus populations were relatively high and significant (0.02 < F ST < 0.06). In contrast, pairwise F ST were low (<0.01) and mostly non significant within both Atlantic and Norwegian groups. However, a weak genetic structure without any clear pattern was observed within both groups, as shown by a few moderately significant F ST. Negative values of F ST have been found indicating a slight sampling bias, and should be considered equal to zero (Balloux and Lugon-Moulin 2002). The MDS illustrated the pattern of genetic differentiation shown with the pairwise F ST comparisons: axes 1 and 2 explained respectively (1) the difference between Norwegian and Atlantic populations, and (2) the difference between the Mediterranean population and all other populations (Fig. 3). Exact tests of genic differentiation showed a similar pattern, with a highly significant differentiation between the Atlantic and Norwegian groups and the Mediterranean population. Interestingly, exact tests showed also a higher genetic structure within the Atlantic cluster, compared to pairwise F ST (Table 1).

The AMOVA revealed that 95.96 % (F it = 0.040) of the genetic variation was explained by variation within individuals, and 3.56 % (F ct = 0.036) by variation between groups (Atlantic vs Norwegian). Both were highly significant (p < 0.001). Variation among populations within groups (F sc = −0.005) and variation among individuals within populations (F IS = 0.010) were not significant. These AMOVA results indicated a global genetic homogeneity both within each population and each group, but a significant genetic differentiation between the Atlantic and Norwegian groups.
Comparisons between the two groups of samples with Fstat showed a significant difference for R a (p = 0.001): Norwegian populations showed, on average, 0.9 less allele per locus than Atlantic populations. A slight but significant difference was also detected for H o (H o-ATL = 0.644, H o-NOR = 0.614, p = 0.03). However, no difference was detected in F IS values. The Mantel test for IBD was significant over the whole dataset (excluding the Mediterranean population) (n = 14, r = 0.83, p = 0.006) and also for the Norwegian group, despite our limited number of samples (n = 6, r = 0.73, p = 0.03). In contrast, no IBD was detected in the Atlantic group (n = 8, r = 0.29, p = 0.202).
Discussion
Genetic variability
Overall, the genetic diversity observed in the present study was comparable to those previously reported using microsatellite markers on P. maximus (Watts et al. 2005; Hold et al. 2012, 2013), but higher than the diversity found with EST-derived microsatellites (Charrier et al. 2012). Levels of genetic variability were also comparable with studies on other scallop species (Sato et al. 2005; Kenchington et al. 2006; Arias et al. 2010; Marín et al. 2012).
Null alleles were detected in three out of twelve loci (PmRM043, PmRM007 and PmRM027). One locus (PmRM043) in particular displayed very large F IS values, which were most likely caused by a high frequency of null alleles. Null alleles are frequent in mollusks (McInerney et al. 2010), and they can significantly bias genetic data, e.g. overestimation of F ST estimates (Chapuis and Estoup 2007). However the population structure analysis performed with and without the three loci affected by null alleles gave comparable results (similar global F ST), suggesting that null alleles did not have any impact on the reliability of our results.
Population structure
Overall, microsatellite data showed that populations of P. maximus are clearly structured into two groups of populations: a Norwegian and an Atlantic group (the latter being composed of populations from Galicia to Ireland). Allelic richness and observed heterozygosities significantly differed between the two groups, with Norwegian populations displaying 0.9 less allele on average than Atlantic populations and 0.03 lower heterozygosity (p = 0.001 and p = 0.03 respectively). This difference could be due to a larger proportion of null alleles in Norwegian populations; however, this seems unlikely given the lack of significant differences in F IS values between both groups (a higher proportion of null alleles in Norwegian populations should induce higher F IS values). The clear structuring between the Norwegian and Atlantic populations is likely to be driven by the observed difference in allelic richness. As a result, the significance of the Mantel test over the whole set of populations might reflect this difference rather than overall IBD (see also below regarding IBD within groups). Although the great scallop can have a long PLD (24–78 days in laboratory conditions; Beaumont and Barnes 1992) and therefore great ability to disperse (O’Connor et al. 2007), the genetic difference between the two groups could result, at least partly, from hydrographic features within the northern North Sea, which may constitute a barrier to dispersal between Norway and the North Atlantic (Lee 1980; Huthnance 1991, 1997; Otto et al. 1990; Hislop et al. 2001). In addition, the Norwegian Trench (100 km across, 300–700 m deep; Huthnance 1991) constitutes a barrier for benthic bivalves such as the great scallop (Rosenberg et al. 1996), thus possibly reducing the connectivity between P. maximus populations from the western and eastern North Sea.
The population structure assessed with microsatellites is consistent with results from previous investigations. Ridgway and Dale (2000) identified a clear differentiation between two Norwegian samples and twelve British and French samples (data from Wilding et al. 1997) using mitochondrial RFLPs. Discrepancies between levels of population differentiation using nuclear and mitochondrial genomes have often been observed in bivalve species (e.g. in the European flat oyster, Diaz-Almela et al. 2004). Differences in effective population size related to the mode of inheritance, sex-dependant variance of reproductive success or sex-biased dispersal modes have been commonly proposed to explain such differences. In the case of the great scallop, no clear difference in terms of genetic differentiation can be noticed between mitochondrial and nuclear markers (allozymes: Beaumont et al. (1993); present microsatellite data). Hermaphroditism in the great scallop might indeed contribute to reduce eventual discrepancies between nuclear and mitochondrial markers. The congruent genetic structures observed between Norwegian and Atlantic populations for both types of markers support their phylogeographic significance.
A large population group with a very limited structure, ranging from the Iberian peninsula to the British Isles, and genetically differentiated from a more northern group, has been observed in other marine invertebrates: the flat oyster Ostrea edulis (Launey et al. 2002), the green crab Carcinus maenas (Roman and Palumbi 2004) and the common cockle Cerastoderma edule (Krakau et al. 2012). In most cases, a phylogeographical explanation has been proposed, suggesting that this genetic structure may originate from the existence of two glacial refugia: one southern, below the glaciated area and source of the Atlantic recolonization, and one northern, source of the Norwegian recolonization. Following this scenario, the genetic structure found in the great scallop would be thus explained by two independent recolonization events. However, in the case of P. maximus, a decreasing genetic diversity with latitude is apparent, thus supporting also the hypothesis of a single southern glacial refugium, with successive post-glacial (re)colonization events occurring firstly in the North-East Atlantic and later along the Norwegian coasts. According to this second hypothesis, the lower genetic diversity of Norwegian populations would thus result from founder effects that may have progressively reduced the diversity during successive (re)colonization events toward northern regions.
Norwegian cluster
The observed genetic structure of populations sampled along the Norwegian coast revealed small but significant IBD but no significant gradient in genetic diversity. Along Norway, currents flow northwards following the coast line, from the South (Skagerrak) up into the Arctic Ocean and the Barents Sea (Hopkins 1991). As a result, it is possible that a gradient of genetic diversity exists but our data is not sufficient to detect it. However, longer PLD might blur such a gradient as it has been shown to be longer at low temperature in the great scallop (Le Pennec et al. 2003).
Atlantic cluster
The weak genetic structure observed within the Atlantic group is less expected than within the Norwegian group. A very low level of differentiation from Spain to the British Isles could result from a lack of power of the microsatellite markers, failing to detect weak signals of genetic differentiation. Alternatively, this limited differentiation among Atlantic populations might be explained by regular (albeit low) gene flow over generations. High gene flow is likely to occur along French and British coasts, as P. maximus populations are found ubiquitously (Brand 2006). However a stepwise migration model is unlikely between Galicia and France, as there is no significant IBD detected among these regions. There is a gap in the geographic range of P. maximus along the northern coast of Spain, as the continental shelf is too narrow (i.e. there is not much area displaying a compatible depth with the maintenance of a functional population of P. maximus) and water temperatures are too high (Brand 2006). However, even though distances among populations are often too large to ensure a high connectivity, larvae could occasionally disperse over broad distances during exceptional events (e.g. wind-forced currents, storms). Genetic differentiation is not only driven by migration (or its lack of), but also by genetic drift. In our study, we could not estimate effective population sizes, most likely because they are too large and/or our markers are not sufficiently informative. So the lack of genetic differentiation along all the Atlantic coasts likely results from a combination of gene flow induced by larval dispersal and low genetic drift linked to large effective population sizes.
Comparison with phenotypic studies
The pattern of genetic structuring found in the present study can also be compared with the phenotypic variability observed in natural populations. Chauvaud et al. (2012) reported differences in shell growth patterns between “Southern” (from Spain to Wales) and “Northern” populations (from Scotland to North of Norway), and hypothesized that it could be due to phenotypic plasticity and/or genetic differentiation. We found a similar pattern of differentiation in this study, but the boundary between the two groups was placed slightly differently. Indeed, the combination of our study with those from Hold (2012) have shown that the “Atlantic” group (genetic-wise) expands from Spain to the northern North Sea. Further inquiries are needed to explore the mismatch between the phenotypic and genetic boundaries.
Similarly, phenotypic differences observed in reproductive strategies between Saint-Brieuc Bay and Scottish populations (Paulet et al. 1988; Cochard and Devauchelle 1993) could also have a genetic basis. However, in our study, The Saint-Brieuc Bay population was not clearly differentiated (even if some F ST, albeit low, were significant). The use of non-neutral markers, identified using a genome-scan approach, could be useful for a further exploration of this question. The proteomic study of Artigaud et al. (2014) revealed a differentiation of proteomes between a Norwegian population and an Atlantic one. These results may partly be explained by the genetic structure observed here, although they are likely to be largely influenced by environmental factors (i.e. phenotypic plasticity).
Genetic differentiation of Mediterranean scallops
Aside from their geographic distribution, the two European scallop species can be distinguished on the basis of their shell morphology (Rombouts 1991). Several genetic studies using allozymes (Rios et al. 2002) or mitochondrial DNA (Wilding et al. 1999; Saavedra and Peña 2004, 2005) have shown very little differentiation between the two taxa, suggesting that the separation of P. maximus and P. jacobaeus into two species may not be justified. Although microsatellites are clearly not appropriate markers to perform phylogenetic studies, it is interesting to note that no cross-species amplification problems and similar level of allelic richness were observed when genotyping Mediterranean samples using loci initially developed on Atlantic scallops. However, a comprehensive study remains to be conducted to explore thoroughly the taxonomic and phylogenetic distinction between P. maximus and P. jacobaeus.
Potential genetic influence of hatchery-based population enhancement in the Bay of Brest
The population of the Bay of Brest has potentially undergone a strong bottleneck effect caused by massive mortality in the winter of 1962–1963 (Dao et al. 1999), and has then been heavily stocked by a sea ranching program for approximately 30 years (Beaumont and Gjedrem 2006; Fleury et al. 2005). Interestingly, we have not detected a lower genetic diversity (at any of the measured parameters) compared with other Atlantic populations, or any obvious alteration of the natural population, contrary to what could possibly be expected as a result of hatchery-based population enhancement (Beaumont and Gjedrem 2006; Gaffney 2006). This is clearly shown by the magnitude of genetic diversity in relation to latitude: anthropogenic pressure has not affected the pattern of diversity resulting from the natural evolutionary history of the population from the Bay of Brest. This observation could be explained by several hypotheses: first, gene flow originating from outside the Bay of Brest may maintain a high level of genetic diversity within the population from the Bay. Alternatively, the reproductive success of hatchery-raised individual could be low in the wild, as seen in salmonids (Araki et al. 2007; Christie et al. 2012; Milot et al. 2013). In particular, seeding sites are obviously known by fishermen in the Bay of Brest, and this may result in a higher fishing pressure on hatchery-produced individuals than on wild individuals, thus drastically reducing their reproductive contribution. Furthermore, in some conditions of seeding, size of hatchery broodstock and reproductive success of hatchery-born individuals, Gaffney (2006) hypothesized an increase in effective population size due to the Ryman-Laikre effect (Ryman and Laikre 1991) in mollusk populations with large effective size in the wild, thus preventing any decrease of the genetic diversity. This effect has been confirmed in P. maximus in the Isle of Man by a simulation study (Hold et al. 2013), and this may be also the case in the Bay of Brest. Finally, the high level of diversity within the population of Brest may result from good aquaculture practices at the Tinduff hatchery. Indeed, the broodstock used in this hatchery is renewed each year by sampling new genitors in the wild. Moreover, to produce the requested number of spat, up to 100-120 genitors can be used in a single year.
Tettelbach et al. (2002) demonstrated that the potential reproductive success of hatchery-born individuals of Argopecten irradians irradians was similar to the potential reproductive success of wild-born individuals, but there was no differential fishing pressure taken into account. Similarly, a lack of impact of aquaculture practices was reported in the Japanese scallop Patinopecten yessoensis (Sato et al. 2005) in Japan, but some were found in hatchery production of the same species in China (Li et al. 2007). However, the magnitude of seeding and the renewal of the broodstock were very different than in the Bay of Brest. Moreover, in our study we sampled only the natural population, at one particular year. Sampling both natural and seeded populations, during several different years, will be necessary to continue exploring in details this question.
Conclusion and perspectives
P. maximus population structure appears to reflect both recent evolutionary history and actual pattern of connectivity: two clusters were observed, possibly originating from one or two glacial refugia. The observed genetic structure might be maintained by barriers to larval dispersal, like the Norwegian trench. This genetic structure does not seem to be impacted by anthropogenic pressure such as fishing and sea-ranching aquaculture. A more comprehensive study of the impact of sea-ranching practices, particularly in the Bay of Brest, is needed to (1) better understand the genetic and demographic interactions between stock enhancement through hatchery-based seeding and natural recruitment and (2) identify eventual adaptive changes related to hatchery-propagation or local adaption in a rapidly changing environment. Analyzing these issues could help developing a valuable model of a responsible management of an important marine resource, possibly transferable to others marine resources in need of better management. Finally, the limited results on the Mediterranean population analyzed in this study highlight the need for a comprehensive comparative study on the genetics of P. maximus and P. jacobaeus.
References
Andersen S, Christophersen G, Magnesen T (2011) Spat production of the great scallop (Pecten maximus): a roller coaster. Can J Zool 89(7):579–598
Araki H, Cooper B, Blouin MS (2007) Genetic effects of captive breeding cause a rapid cumulative fitness decline in the wild. Science 318(5847):100–103
Arias A, Freire R, Méndez J, Insua A (2010) Isolation and characterization of microsatellite markers in the queen scallop Aequipecten opercularis and their application to a population genetic study. Aquat Living Resour 23(2):199
Artigaud S, Lavaud R, Thébault J, Jean F, Strand Ø, Strohmeier T, Pichereau V (2014) Proteomic-based comparison between populations of the Great Scallop Pecten maximus. J Proteomics 105:164–173
Balloux F, Lugon-Moulin N (2002) The estimation of population differentiation with microsatellite markers. Mol Ecol 11(2):155–165
Beaumont A (2000) Genetic considerations in transfers and introductions of scallops. Aquac Int 8(6):493–512
Beaumont A, Barnes DA (1992) Aspects of veliger larval growth and byssus drifting of the spat of Pecten maximus and Aequipecten (Chlamys) opercularis. ICES J Mar Sci 49:417–423
Beaumont A, Gjedrem T (2006) Scallops—Pecten maximus and P jacobaeus. Genetic impact of aquaculture activities on native populations. Genimpact final scientific report pp 83–90
Beaumont A, Morvan C, Huelvan S, Lucas A, Ansell AD (1993) Genetics of indigenous and transplanted population of Pecten maximus—no evidence for the existence of separate stocks. J Exp Mar Biol Ecol 169:77–88
Belkhir K, Borsa P, Goudet J, Chikhi L, Bonhomme F (2001) GENETIX Logiciel Sous WindowsTM Pour La Génétique Des Populations. Laboratoire Génome, Populations, Interactions CNRS UMR 5000, Université de Montpellier II, Montpellier (France)
Boudry P, Collet B, Cornette F, Hervouet V, Bonhomme F (2002) High variance in reproductive success of the Pacific oyster (Crassostrea gigas Thunberg) revealed by microsatellite-based parentage analysis of multifactorial crosses. Aquaculture 204(3):283–296
Brand AR (2006) Scallop ecology: distributions and behavior. Dev Aquac Fish Sci 35:651–744
Chapuis MP, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24(3):621–631
Charrier G, Morvezen R, Calves I, Laroche J (2012) Development of new microsatellite markers derived from expressed sequence tags for the great scallop (Pecten maximus). Conserv Genet Resour 4(4):931–934
Chauvaud L, Patry Y, Jolivet A, Cam E, Le Goff C, Strand O, Charrier G, Thebault J, Lazure P, Gotthard K, Clavier J (2012) Variation in Size and Growth of the Great Scallop Pecten maximus along a latitudinal gradient. PLoS ONE 7:e37717
Christie MR, Marine ML, French RA, Blouin MS (2012) Genetic adaptation to captivity can occur in a single generation. Proc Natl Acad Sci 109(1):238–242
Cochard JC, Devauchelle N (1993) Spawning fecundity and larval survival and growth in relation to controlled conditioning in native and transplanted populations of Pecten maximus (L): evidence for the existence of separate stocks. J Exp Mar Biol Ecol 169(1):41–56
Dao JC, Buestel D, Gerard A, Halary C, Cochard JC (1985) Le programme de repeuplement de coquille Saint-Jacques: finalité résultats et perspectives. In: IV ème Colloque Franco-Japonais: Les aménagements côtiers et la gestion du littoral
Dao JC, Fleury PG, Barret J (1999) Scallop culture in Europe. In: Howell B, Moksness E, Svåsand T (eds) Stock Enhancement and Sea Ranching. Fishing News Books, Blackwell Science, pp 423–4435
Diaz-Almela E, Boudry P, Launey S, Bonhomme F, Lapegue S (2004) Reduced female gene flow in the European flat oyster Ostrea edulis. J Hered 95(6):510–516
Excoffier L, Lischer HE (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10(3):564–567
FAO (2015) Global Capture Production. http://www.faoorg/fishery/statistics/global-capture-production/en. Accessed 23 February 2015
Fleury PG, Carval JP, Muzellec ML, Gerard A, Barret J, Cochard JC, Dao JC (2005) Histoire D’une Complémentarité Entre L’aquaculture et La Pêche: 20 Ans de Production de Coquilles Saint-Jacques D’écloserie Pour Semis Sur Zones de Pêche: Rade de Brest 1983–2002. Report available at http://www.archimerifremerfr/doc/00000/7308/
Gaffney PM (2006) The role of genetics in shellfish restoration. Aquat Living Resour 19:277–282
Goudet J (1995) FSTAT (Version 12): a computer program to calculate f-statistics. J Hered 86(6):485–486
Heipel DA, Bishop JDD, Brand AR, Thorpe JP (1998) Population genetic differentiation of the great scallop Pecten maximus in western Britain investigated by randomly amplified polymorphic DNA. Mar Ecol Prog Ser 162:163–171
Heipel DA, Bishop JDD, Brand AR (1999) Mitochondrial DNA variation among open-sea and enclosed populations of the scallop Pecten maximus in western Britain. J Mar Biol Assoc U K 79:687–695
Hislop JR, Gallego A, Heath MR, Kennedy FM, Reeves SA, Wright PJ (2001) A synthesis of the early life of the anglerfish Lophius piscatorius (Linnaeus 1758) in northern British waters. ICES J Mar Sci 58:70–86
Hold N (2012) An investigation into the spatial scales of genetic and reproductive variation in the scallop Pecten maximus L. Doctoral dissertation, Bangor University
Hold N, Murray LG, Kaiser MJ, Hinz H, Beaumont AR, Taylor MI (2012) Potential effects of stock enhancement with hatchery-reared seed on genetic diversity and effective population size. Can J Fish Aquat Sci 70(2):330–338
Hold N, Dawnay L, Taylor MI (2013) Development of microsatellite markers from 454 transcriptome derived sequences for the scallop Pecten maximus. Conserv Genet Resour 5(3):663–666
Hopkins TS (1991) The GIN Sea—A synthesis of its physical oceanography and literature review 1972–1985. Earth Sci Rev 30(3):175–318
Huthnance JM (1991) Physical oceanography of the North Sea. Ocean Shorel Manag 16(3):199–231
Huthnance JM (1997) North Sea interaction with the North Atlantic ocean Dtsch Hydrogr Z 49(2–3):153–162
Kenchington EL, Patwary MU, Zouros E, Bird CJ (2006) Genetic differentiation in relation to marine landscape in a broadcast spawning bivalve mollusc (Placopecten magellanicus). Mol Ecol 15(7):1781–1796
Kitada S, Kitakado T, Kishino H (2007) Empirical Bayes inference of pairwise FST and its distribution in the genome. Genetics 177(2):861–873
Krakau M, Jacobsen S, Jensen KT, Reise K (2012) The cockle Cerastoderma edule at Northeast Atlantic shores: genetic signatures of glacial refugia. Mar Biol 159(1):221–230
Launey S, Ledu C, Boudry P, Bonhomme F, Naciri-Graven Y (2002) Geographic structure in the European flat oyster (Ostrea edulis L) as revealed by microsatellite polymorphism. J Hered 93(5):331–351
Le Pennec M, Paugam A, Pennec GL (2003) The pelagic life of the pectinid Pecten maximus—a review. ICES J Mar Sci J Cons 60:211–233
Lee AJ (1980) North Sea: physical oceanography. Elsevier Oceanogr Ser 24:467–493
Li Q, Xu K, Yu R (2007) Genetic variation in Chinese hatchery populations of the Japanese scallop (Patinopecten yessoensis) inferred from microsatellite data. Aquaculture 269(1–4):211–219
Ma XL, Cowles DL, Carter RL (2000) Effect of pollution on genetic diversity in the bay mussel Mytilus galloprovincialis and the acorn barnacle Balanus glandula. Mar Environ Res 50(1):559–563
Mackie LA, Ansell AD (1993) Differences in reproductive ecology in natural and transplanted populations of Pecten maximus: evidence for the existence of separate stocks. J Exp Mar Biol Ecol 169(1):57–75
Magnesen T, Christophersen G (2008) Reproductive cycle and conditioning of translocated scallops (Pecten maximus) from five broodstock populations in Norway. Aquaculture 285:109–116
Marín A, Fujimoto T, Arai K (2012) Isolation and characterization of 12 microsatellite loci in the Peruvian scallop Argopecten purpuratus and cross-species amplification in other scallop species (family Pectinidae). Conserv Genet Resour 4(1):179–182
McInerney CE, Allcock AL, Johnson MP, Bailie DA, Prodöhl PA (2010) Comparative genomic analysis reveals species-dependent complexities that explain difficulties with microsatellite marker development in mollusks. Heredity 106(1):78–87
Milot E, Perrier C, Papillon L, Dodson JJ, Bernatchez L (2013) Reduced fitness of Atlantic salmon released in the wild after one generation of captive breeding. Evol Appl 6(3):472–485
Minchin D (2003) Introductions: some biological and ecological characteristics of scallops. Aquat Living Resour 16(6):521–532
Morvezen R, Cornette F, Charrier G, Guinand B, Lapegue S, Boudry P, Laroche J (2013) Multiplex PCR sets of novel microsatellite loci for the great scallop Pecten maximus and their application in parentage assignment. Aquat Living Resour 26(03):207–213
Nicolle A, Dumas F, Foveau A, Foucher E, Thiébaut E (2013) Modelling larval dispersal of the king scallop (Pecten maximus) in the English Channel: examples from the bay of Saint-Brieuc and the bay of Seine. Ocean Dyn 63(6):661–678
O’Connor MI, Bruno JF, Gaines SD, Halpern BS, Lester SE, Kinlan BP, Weiss JM (2007) Temperature control of larval dispersal and the implications for marine ecology evolution and conservation. Proc Natl Acad Sci 104(4):1266–1271
Otto L, Zimmerman JTF, Furnes GK, Mork M, Saetre R, Becker G (1990) Review of the physical oceanography of the North Sea. Neth J Sea Res 26(2):161–238
Paulet YM, Lucas A, Gerard A (1988) Reproduction and larval development in two Pecten maximus (L) populations from Brittany. J Exp Mar Biol Ecol 119(2):145–156
R Core Team (2012) R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna Austria ISBN 3-900051-07-0. http://www.R-project.org/
Ridgway GMI, Dahle G (2000) Population genetics of Pecten maximus of the Northeast Atlantic coast. Sarsia 85:167–172
Rıos C, Sanz S, Saavedra C, Pena JB (2002) Allozyme variation in populations of scallops Pecten jacobaeus (L) and P. maximus (L) (Bivalvia: Pectinidae) across the Almeria–Oran front. J Exp Mar Biol Ecol 267(2):223–244
Robert R, Gérard A (1999) Bivalve hatchery technology: the current situation for the Pacific oyster Crassostrea gigas and the scallop Pecten maximus in France. Aquat Living Resour 12(02):121–130
Roman J, Palumbi SR (2004) A global invader at home: population structure of the green crab Carcinus maenas in Europe. Mol Ecol 13(10):2891–2898
Rombouts A (1991) Guidebook to Pecten Shells. Crawford House Press, Bathurst
Rosenberg R, Hellman B, Lundberg A (1996) Benthic macrofaunal community structure in the Norwegian Trench deep Skagerrak. J Sea Res 35(1):181–188
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8(1):103–106
Ryman N, Laikre L (1991) Effects of supportive breeding on the genetically effective population size. Conserv Biol 5:325–329
Saavedra C, Peña JB (2004) Phylogenetic relationships of commercial European and Australasian king scallops (Pecten spp) based on partial 16S ribosomal RNA gene sequences. Aquaculture 235(1):153–166
Saavedra C, Peña JB (2005) Nucleotide diversity and Pleistocene population expansion in Atlantic and Mediterranean scallops (Pecten maximus and P. jacobaeus) as revealed by the mitochondrial 16S ribosomal RNA gene. J Exp Mar Biol Ecol 323(2):138–150
Sato M, Kawamata K, Zaslavskaya N, Nakamura A, Ohta T, Nishikiori T, Nagashima K (2005) Development of microsatellite markers for Japanese scallop (Mizuhopecten yessoensis) and their application to a population genetic study. Mar Biotechnol 7(6):713–728
Slatkin M (1995) A measure of population subdivision based on microsatellite allele frequencies. Genetics 139(1):457–462
Smith PJ, Francis RICC, McVeagh M (1991) Loss of genetic diversity due to fishing pressure. Fish Res 10(3):309–316
Tettelbach ST, Smith CF, Wenczel P, Decort E (2002) Reproduction of hatchery-reared and transplanted wild bay scallops Argopecten irradians irradians relative to natural populations. Aquac Int 10(4):279–296
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary N-E using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3(3):244–262
Watts PC, Mallanaphy PJ, Mccarthy C, Beukers-Stewart BD, Mosley MW, Brand AR, Saccheri IJ (2005) Polymorphic microsatellite loci isolated from the great scallop Pecten maximus (Bivalvia: Pectinidae). Mol Ecol Notes 5(4):902–904
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370
Wilding CS, Beaumont AR, Latchford JW (1997) Mitochondrial DNA variation in the scallop Pecten maximus (L) assessed by a PCR-RFLP method. Heredity 79:178–189
Wilding CS, Beaumont AR, Latchford JW (1999) Are Pecten maximus and Pecten jacobaeus different species? J Mar Biol Assoc UK 79:949–952
Acknowledgments
We would like to thank the two anonymous reviewers for insightful comments and suggestions. We would also like to thank all people involved in sampling: Florence Cornette, Romuald Massé, Jose Fuentes, Julien Normand, Romain Lavaud, Michael Retho, Arne Duinker, Geir Askvik Haugum and Tore Strohmeier. We also thank the Tinduff hatchery for a fruitful cooperation with our laboratory, particularly Jean-Pierre Carval and Marie-Louise Muzellec. We are also grateful to Edouard Lavergne for his help in data analysis and for providing the MDS R script.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Morvezen, R., Charrier, G., Boudry, P. et al. Genetic structure of a commercially exploited bivalve, the great scallop Pecten maximus, along the European coasts. Conserv Genet 17, 57–67 (2016). https://doi.org/10.1007/s10592-015-0760-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-015-0760-y