
Abstract
The way that genetic variation is distributed geographically has important conservation and evolutionary implications. Here, we examined the distribution of genetic variation within and among populations of the montane perennial Ipomopsis aggregata. We sampled plants in western Colorado and examined (1) population genetic structure over a geographic area that spanned 130 km, including genetic variation within disturbed and undisturbed sites; (2) the relationship between genetic differentiation and geographic distance; and (3) the relationship between flowering time and genetic differentiation among plants within and among geographic areas. F IS was significantly higher (t test, P = 0.006), expected heterozygosity was significantly lower (t test, P = 0.04), and allelic richness was marginally significantly lower (t test, P = 0.078) among anthropogenically-disturbed sites compared to undisturbed sites. We found moderate genetic differentiation over the area sampled (average pairwise F ST = 0.04; average pairwise \(F^{\prime}_{ST} = 0.19\)), but no association of genetic and geographic distance (Mantel test P values 0.44 for F ST and 0.36 for \(F^{\prime}_{ST}\)). We found a strong association of flowering time and genetic differentiation over small and large spatial scales. Genetic differentiation between early and late flowering plants within a focal site was statistically significant (genic test for population differentiation combined P value <0.001; F ST = 0.05). There was a significant correlation between genetic distance (\(F^{\prime}_{ST}\)) and distance in flowering time, when controlling for geographic distance, over the whole geographic area (Partial Mantel test R xy = 0.32, P = 0.013). A multiple regression with randomization further supported the inference that flowering time, but not geographic distance or elevation, predicted \(F^{\prime}_{ST}\) (geographic distance: β = −0.03, P = 0.89; elevation: β = 0.01, P = 0.96; phenological distance: β = 0.30, P = 0.05), but not F st (geographic distance: β = −0.02, P = 0.92; elevation: β = 0.14, P = 0.38; phenological distance: β = 0.25, P = 0.11), unless elevation was left out of the model (geographic distance: β = −0.03, P = 0.9; phenological distance: β = 0.29, P = 0.03). The association of flowering time and genetic distance despite the lack of isolation by distance provides further evidence for the usefulness of incorporating this variable into plant landscape genetic studies when possible.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The way that genetic variation is distributed geographically has important evolutionary and conservation implications (Allendorf and Luikart 2007; Frankham 2010). Analyses of population genetic structure can help identify where connectivity might have been decreased by human activities as well as reveal small, genetically depauperate populations that might have elevated probabilities of inbreeding depression and lower probabilities of persistence (Soulé 1987; Gilpin and Hanski 1991; Saccheri et al. 1998). The geographic distribution of genetic variation within networks of connected and locally adapted populations also shapes the way species respond to environmental changes (Jay et al. 2012), for example through what has been termed the Portfolio effect in which genetic diversity within and divergence among connected populations enhances long-term sustainability of populations experiencing environmental changes (Shindler et al. 2010).
The field of landscape genetics combines population genetics with landscape ecology to explain how landscape features—such as rivers or mountains—influence the distribution of genetic variation (Manel et al. 2003; Storfer et al. 2007; Holderegger and Wagner 2008). In addition to geographic distance (i.e., isolation by distance, IBD; Wright 1943) or environmental characteristics such as elevation or habitat type (i.e., isolation by environment, IBE; Wang 2013), an important, but less often considered factor spatially shaping intraspecific genetic structure is the timing of reproductive events (Stanton et al. 1997). Reproductive timing determines mate availability, so individuals that reproduce at more similar times will be more likely to mate with one another. If a population contains individuals that reproduce at heritably different times, genetic differentiation among individuals should be positively related to increasing differences in reproductive timing, a phenomenon called ‘isolation by time’ by Hendry and Day (2005). Hendry and Day (2005) predict that increasing heritabilities of reproductive time will lead to a stronger association of differences in reproductive time and genetic differences. Whether or not reproductive timing is heritable, it is plausible that individuals could show an association between phenology and relatedness if reproductive timing is governed by environmental variables that vary spatially in a consistent manner, and if dispersal is restricted.
For plants, associations between flowering time and genetic structure are likely because plants that flower at the same time have the opportunity to exchange pollen. Though pollen and seed movement both contribute to gene flow in plant populations, pollen movement is usually more important in genetically structuring populations (Levin and Kerster 1974; Fenster 1991; Ennos 1994). Theoretical (Fox 2003) and empirical (Weis and Kossler 2004) work suggests that plants mate assortatively based on their flowering time. Flowering time may be influenced both by genetic and environmental factors, and the relative strengths of these likely vary across species. Flowering time is highly heritable for some plants (reviewed in Geber and Griffen 2003), under strong selection (Korves et al. 2007; Colautti and Barrett 2013), and selection on flowering time can lead to genetic differentiation among plants that are subject to different selection regimes (Kittelson and Maron 2001; Hall and Willis 2006). Flowering time is also influenced by environmental conditions such as light and nutrient level (Stanton et al. 2000), and the timing of snowmelt (Inouye and McGuire 1991; Price and Waser 1998; Dunne et al. 2003; Lambert et al. 2010).
Spatial heterogeneity in environmental conditions could lead to an association between genetic differentiation and flowering time. Flowering time is often associated with genetic clines at the continental scale, presumably due to selection regimes that vary latitudinally (Stinchcomb et al. 2004; Keller et al. 2011). At more local spatial scales of less than tens of kilometers, associations between flowering time and patterns of genetic differentiation are mixed. Given the tendency of insects (Levin and Kerster 1974) and hummingbirds (Waser and Price 1983) to move pollen among plants in close proximity, genetic differences could build up over time among plants in sites that differ consistently in the timing of snowmelt, especially for species in which seed dispersal is limited. Some studies have found associations of flowering time and patterns of genetic differentiation that are likely due to differences in the timing of snowmelt (Hirao and Kudo 2004; Yamagashi et al. 2005; Hirao and Kudo 2008). Shimono et al. (2009) did not explicitly examine how flowering time influences genetic structure, but did find that variation in snowmelt explained patterns of genetic differentiation. On the other hand, if seed movement influences gene flow more than pollen movement, flowering time may be associated with date of snowmelt, but gene flow may not be significantly restricted between sites that flowered at different times (Stanton et al. 1997; Gerber et al. 2004; Cortés et al. 2014). Given widespread climate-changed induced phenological shifts (Parmesan and Yohe 2003), studies that investigate the extent to which genetic structure is associated with flowering time will be important for informing species-specific predictions of how the geographic distribution of genetic variation may change under future climate change scenarios.
Here, we examined the distribution of genetic variation within and among populations of Ipomopsis aggregata (Polymoniaceae), a self-incompatible, monocarpic, montane perennial with limited seed dispersal (Waser and Price 1983). First, we investigated genetic structure over a geographic area that spanned 130 km, and determined how genetic diversity differed among populations that varied in anthropogenic influence. Second, we explored the relationship between genetic distance and geographic distance, and landscape characteristics and genetic distance. Third, we explored the relationship between flowering time and genetic distance over both small (50 m scale) and large (130 km) spatial scales.
Methods
Species and sampling
Ipomopsis aggregata is found in diverse montane habitats of the western United States. It is monocarpic, self incompatible, and grows as a hardy rosette for several years until a flowering stalk is produced, after which reproduction and then death occur. Individual flowers are open for about 3–5 days and plants flower for about 4–6 weeks (Freeman et al. 2003). The flowers are red, tubular, protandrous, and are on paniculate-racemose inflorescences. The seeds of Ipomopsis aggregata fall near the parent plant (Waser and Price 1983), so gene flow is thought to occur primarily via transfer of pollen by pollinators. Common visitors include broad-tailed and rufous hummingbirds (Selasphorus platycercus and S. rufus), and also occasionally bumble bee queens (Bombus appositus), some solitary bees, butterflies, hawkmoths, and flies.
In June and July 2011 we sampled tissue from 297 Ipomopsis aggregata plants in the greater geographic area around the Rocky Mountain Biological Laboratory in Gothic, CO (Fig. 1). We sampled on average 20 plants at 14 sites (Table 1), in sites along the main drainage that runs from Gunnison to Crested Butte, and up tributaries to this drainage. The 14 sites were separated by 2.6–130 km. Within each sampling location we sampled plants within a circle of radius 50 m. Some sites consisted of small, isolated populations; these tended to be along roads and in disturbed areas while others were in more natural meadows, in which I. aggregata plants extended beyond the 50 m radius. Ipomopsis aggregata plants are sometimes grazed by deer and elk; we included only ungrazed plants in this study. We removed one leaf from each sampled plant, and subsequently dried leaf tissue for storage until genetic analysis. We sampled only from flowering plants, and it is likely that some of these plants germinated in different years (Waser and Price 1989). For one of these 14 sites (hereafter called our focal site; site 4 in Table 1), we conducted detailed observations of flowering time for individual plants in a meadow (see below for details).

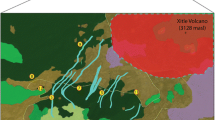
Map of sampling locations in which sites are represented by circles, and colors represent the genetic groups to which individuals within each site were assigned based on STRUCTURE results using K = 3 and a prior that takes sampling location into account
Molecular analyses
We genotyped plants at eight microsatellite loci developed for I. aggregata: P4, P7, P8, P9, P109, P121, P166, and P186 (Wu 2006), after extracting DNA with a standard salt precipitation procedure. We amplified seven primers in one multiplex PCR reaction, and we amplified P109 by itself. The PCR reaction conditions were as follows: 94 °C for 15 min, followed by 35 cycles of 94 °C for 30 s, 57 °C for 90 s, 72 °C for 60 s, followed by 72 °C for 30 min. Following PCR we ran samples on a 3730 DNA sequencer (Applied Biosystems) in the Genomics Core Facility at the University of Arizona, and analyzed the microsatellite lengths using GENEMAPPER (Applied Biosystems) and Geneious software v7.0 (Biomatters Ltd, Auckland, New Zealand). All samples were scored twice.
Genetic differentiation
We calculated levels of genetic diversity within sampling locations (mean number of alleles and heterozygosity), and tested for departure from Hardy–Weinberg (HW) proportions using the program Genalex (Peakall and Smouse 2006). We used the R package Hierfstat to calculate allelic richness, and tested for linkage disequilibrium among pairs of loci using Genepop (Rousset 2008). To correct for inflated type I error rates due to multiple testing we used a sequential Bonferroni correction, at an alpha level of 0.05, within each population. We tested for departure from HW proportions separately for “early” and “late” flowering plants on our focal site because we found significant genetic differentiation between these two groups (see “Results” section). We used Genalex to calculate F IS averaged over loci for each site, and conducted a t test to test for possible differences in F IS, heterozygosity, and allelic richness between sites in which populations were in disturbed areas along trail or roadsides and those in more natural meadows in which the population of I. aggregata plants expanded beyond the area of 50 m radius in which we sampled.
To estimate genetic differentiation among sites, we used Nei’s unbiased estimator of G ST (Nei 1987) as an estimate of F ST (Wright 1951) and \(G^{\prime\prime}_{ST}\) (Miermans and Hedrick 2011) as an estimate of \(F^{\prime}_{ST}\), using the program Genodive (Meirmans and Van Tienderen 2004). \(F^{\prime}_{ST}\) was developed to address the fact that \(F_{{ST^{ - } }}\) like estimators are biased low when heterozygosity is high (Jost 2008). \(G^{\prime\prime}_{ST}\) is a standardized \(F^{\prime}_{ST}\) estimator that takes heterozygosity within populations into account and corrects for a small number of sampled populations (Miermans and Hedrick 2011). We tested for locus-specific allele frequency (genic) differences among sampling locations with exact tests implemented with Genepop and combined locus-specific tests with Fisher’s method (Ryman et al. 2006). We estimated the overall statistical significance of the multiple comparisons among sampling locations by employing the B-Y false discovery rate (FDR) method (Benjamini and Yekutieli 2001), which corrects for increased type I error while also controlling the level of falsely rejected null hypotheses (Narum 2006). Finally, we used principal components analysis to examine genetic differences among sites. We used site-specific allele frequencies and specified a covariance matrix with Genodive.
We also used a Bayesian clustering method to detect population structure. STRUCTURE version 2.3.2 (Pritchard et al. 2000) estimates the number of populations (K) and assigns individuals to populations. We ran STRUCTURE under the admixture model, with correlated allele frequencies, using the λ = 1 option. λ parameterizes the allele frequency prior with a Dirichlet distribution of allele frequencies. In addition, we used the “infer ALPHA” option, where ALPHA is the Dirichlet parameter for degree of admixture. We first varied burn-in length and number of iterations to check the consistency of results, including one longer run with a burn-in of 200,000 followed by 1,000,000 iterations. It was apparent that a burn in of 20,000 iterations was sufficient to achieve convergence so we then performed eight runs with a burn-in of 20,000 followed by 100,000 iterations, under both the no LOCPRIOR and under the LOCPRIOR model. The LOCPRIOR option uses location information in the prior, and can be helpful when attempting to detect subtle population structure (Hubisz et al. 2009). We evaluated the parameter r from the STRUCTURE models that used a location prior, which parameterizes the amount of information contributed by sample locations (Hubisz et al. 2009). When r is close to or <1, locations are informative. Larger values of r indicate the either locations are not informative or that genetic structure is not correlated with sample locations. To infer the K value best fitting the data, we used the program Structure Harvester (Earl and vonHoldt 2012), which allows the output of STRUCTURE to be visualized and implements the ad hoc ΔK method (Evanno et al. 2005).
Landscape genetics
To investigate how distance and landscape features influence the geographic distribution of genetic variation we conducted a series of Mantel tests, implemented using GenAlEx. First, we estimated the magnitude of isolation by distance by testing the correlation between geographic distance and F ST and \(F^{\prime}_{ST}\). Second, after using STRUCTURE (described above) to assign individuals to genetic groups, we qualitatively matched the resulting boundaries among groups to physical aspects of the landscape. Third, we investigated the extent to which elevation is associated with genetic differentiation. We tested the extent to which (1) the difference in the elevation of pairs of sites; and (2) the cumulative elevation change among pairs of sites was associated with genetic differentiation. We calculated the cumulative elevation change between each pair of sampling locations using the path tool available in Google Earth version 7.0.
Relationship between flowering time and genetic structure
We tested the extent to which genetic differentiation is associated with flowering time in two ways. First, within our focal site we marked I. aggregata plants before they had flowered. We then conducted observations every other day beginning on June 20, and recorded for each plant the day that flowering began. Because flowering time was somewhat episodic on this site (see Online Resource 8), we subsequently divided plants into groups based on when they first had an open flower, and designated two groups of 19 plants, an “early” and “late” group, between which there was at least a 1 week difference in date of first flowering. These two groups were intermixed geographically over the focal meadow, and there was overlap in the flowering time of these groups, such that the plants in the first flowering group were still flowering when the plants in the second flowering group began flowering. We calculated F ST and \(F^{\prime}_{ST}\) between the two flowering groups.
Second, for each site, we sampled leaf tissue from plants on the day that they first flowered. To ensure we accurately recorded the date of first flowering we visited sites periodically until we saw the consistent morphological changes that I. aggregata undergoes before flowering; unopened buds elongate and usually open the following day (Freeman et al. 2003). We did this for 13 of the 14 sites. However, one site was already in full flower when we sampled it, and had no plants with unopened buds, so this site was excluded from analyses that included flowering time (Site 5; see “Results” section). After pinpointing the date of first flowering for a site, we sampled only from plants that had elongated buds, and sampled within 3 days of the date of first flowering. We created a distance matrix that contained the number of days apart that each pair of sites had their first flowers, hereafter ‘phenological distance’. We then conducted a Mantel test to test the correlation between genetic distances F ST and \(F^{\prime}_{ST}\), and phenological distance. We conducted partial Mantel tests implemented using the R statistical language (R development core team 2008), and using the Vegan package (Oksanen et al. 2011) to control for geographic distance while testing for the correlation of genetic distances and phenological distance. We also determined the correlation between flowering date and elevation, and conducted partial Mantel tests to test the correlation of flowering time and genetic distance while controlling for elevation.
Mantel tests are one of the most common methods for assessing the significance of the correlation between matrices and have been widely applied in landscape genetic analyses. However, there has been recent controversy over their use due to low power and high Type I error rates (Raufaste and Rousset 2001; Legendre and Fortin 2010; Miermans 2015). Therefore, we also used a multiple regression approach with a randomized permutation procedure (MMRR) that accounted for the non-independence of variables, as described in Wang (2013). This procedure may be preferable to Mantel tests because of more appropriate Type I error rates, and because it ranks variables in terms of their relative effects on genetic distance (Wang 2013). We used MMRR implemented in R using the function from Wang (2013), with 10,000 permutations, to determine the independent contribution of geographic and phenological distance to variation in genetic distances F ST and \(F^{\prime}_{ST}\). We also incorporated the measure of elevational distance among sites that had the highest Mantel correlation with genetic distance (difference in elevation among sites; see “Results” section) into the model used to predict genetic distance.
Results
Genetic variation within populations
Mean number of alleles ranged from 5.5 to 13.3 (mean = 9.3, SD = 2.2) expected heterozygosity ranged from 0.69 to 0.83 (mean = 0.77, SD = 0.02), and allelic richness ranged from 7.3 to 10 (mean = 9.1, SD = 0.06; Table 1). Prior to correction for multiple tests, 35 (29 %) of all tests for departure from HW expectations were significant (P = 0.05). After correction for multiple tests using a Bonferroni procedure, 16 (13 %) of all tests were significant (Table 1; Online Resource 1). Deviations from HW expectations corresponded to a deficit of heterozygotes within some sites; mean F IS ranged from 0.002 to 0.29 (Table 1). Some loci tended to deviate from HW proportions more than others but a stronger pattern consisted of multiple loci deviating from HW proportions within certain populations (Online Resource 1). Prior to correcting for multiple tests for linkage disequilibrium, 17 (6 %) were significant before and three (1 %) were significant after a sequential Bonferroni correction. F IS was significantly higher (t test, P = 0.006), expected heterozygosity was significantly lower (t test, P = 0.04), and allelic richness was marginally significantly lower (t test, P = 0.078) among disturbed sites along trail or roadsides than among sites in which the I. aggregata expanded beyond the area (circle of radius 50 m) in which we sampled.
Genetic differentiation among populations
There was moderate genetic differentiation among some pairs of sites. Pairwise F st values ranged from 0 to 0.13 (average 0.04) and pairwise \(F^{\prime}_{ST}\) values ranged from 0 to 0.53 (average 0.19; Table 2). All but three of the 91 genic tests of allele frequency differences among pairs of populations were significant before correcting for type I error. After correcting for type I error, 82 (90 %) of the genic tests were significant. The STRUCTURE results revealed that there were two or three distinct genetic groups (K = 2 or 3), when we used sampling location as a prior, but without the location prior, K = 1 had the strongest support (Online Resource 2–4). When we used the LOCPRIOR model, values of the r parameter were <1, which is consistent with location information being informative in delineating groups. For K = 3, the first cluster (red; Fig. 1) contained sites that were located in the main drainage that runs from Gunnison to Crested Butte. The second cluster (green; Fig. 1) contained sites that were more peripheral to this core drainage. The third cluster (blue; Fig. 1) contained primarily one site (site 13) in the main drainage that was completely surrounded by trees. For K = 2, results were similar except that site 13 grouped with the peripheral sites (Online Resource 4).
It is possible that drift caused allele frequencies to converge in the peripheral sites and was responsible for these STRUCTURE results. However, there was little evidence that enhanced drift drove this pattern. Allelic Richness and the proportion of the genome assigned to the second genetic group (q value for K = 3 with LOCPRIOR) were not correlated (r = 0.09, P = 0.41). There was some evidence for clustering of peripheral sites with greater F IS values, and therefore possibly with greater inbreeding levels. There was no correlation between q values and the number of significant departures from Hardy–Weinberg equilibrium per population after a sequential Bonferroni correction at a significance level of α = 0.05 (r = 0.2, P = 0.52). Mean F IS and q values were not correlated (r = 0.26, P = 0.37; Online Resource 5), however, with a peripheral site that had unusually low F IS removed (site 14; Online Resource 6), F IS and q values were significantly correlated (r = 0.62, P = 0.02). In particular, two sites outside of the core drainage appear to be driving the pattern (sites 1 & 2; Table 1).
Additional evidence supports the STRUCTURE clusters. First, the STRUCTURE results were mirrored by the results of a principal component analysis (Online Resource 7). Second, a significant positive correlation between pairwise \(F^{\prime}_{ST}\) values and difference in the average proportion of the genome (q value) of individuals within sites that was assigned to the first STRUCTURE genetic group (Mantel test R xy = 0.55, P = 0.001) demonstrates a close correspondence between \(F^{\prime}_{ST}\) and the STRUCTURE results.
There was no evidence of isolation by distance over the area sampled. Geographic distance and F st (Mantel test R xy = 0.01, P = 0.44) or \(F^{\prime}_{ST}\) (Mantel test R xy = 0.08, P = 0.36) were not significantly correlated. We also found no evidence that elevation was associated with genetic structure of I. aggregata. There was no significant correlation of F ST or \(F^{\prime}_{ST}\) and cumulative difference in elevation among pairs of sites (Mantel test R xy = −0.05, P = 0.48 for F ST ; R xy = 0.02; P = 0.43 for \(F^{\prime}_{ST}\); range of cumulative elevation change distances was 866–20,072 m) or of difference in elevation among pairs of sites (Mantel test R xy = 0.19; P = 0.10 for F ST ; R xy = 0.07; P = 0.33 for \(F^{\prime}_{ST}\)). Three sites comprised the three pairs of sites that were the most genetically similar but they were not geographically adjacent. These were separated by 28, 24, and 9.4 km, and had cumulative elevation changes of 2252, 3341, and 2737 m, but were located along the one main drainage in the geographic area sampled.
Association of flowering time and genetic structure
We found a strong association between flowering time and genetic structure in I. aggregata. Within our focal meadow, the plants we marked began to flower on June 20, and individual plants began flowering into July (Online Resource 8). There was moderate genetic differentiation between plants in our “early” flowering group—those that flowered on or before June 23, and plants in our “late” flowering group—those that flowered on or after June 30 (F ST = 0.05; \(F^{\prime}_{ST} = 0.23\); genic test for population differentiation combined P value <0.001). Across all populations, sites at higher altitudes tended to flower later in the summer (correlation between flowering time and elevation = 0.66, P = 0.01). Notably, one of the sampling locations that STRUCTURE identified as being genetically distinct (Site 14) was the last to begin flowering, which occurred 36 days after a flower opened at the earliest-flowering sampling location. Another genetically distinct location also flowered late with respect to the other sampling locations; Site 13 was the 11th out of 14 sites to begin flowering.
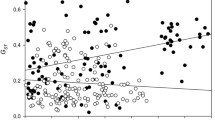
Results from Mantel tests and MMRR analysis indicated that flowering time was associated with genetic structure. There was a significant correlation between phenological distance (defined as difference in flowering time) and F ST (Mantel test R xy = 0.29, P = 0.015) and \(F^{\prime}_{ST}\) (Mantel test R xy = 0.32, P = 0.009; Fig. 2). When controlling for geographic distance there was a significant correlation between phenological distance and F ST (Partial Mantel test R xy = 0.31, P = 0.025), and \(F^{\prime}_{ST}\) (Partial Mantel test R xy = 0.32, P = 0.013). When controlling for elevation there was a significant correlation between phenological distance and F ST (Partial Mantel test R xy = 0.24, P = 0.047), and \(F^{\prime}_{ST}\) (Partial Mantel test R xy = 0.30, P = 0.018). From the MMRR used to predict genetic distance F ST from geographic and phenological distances, phenological distance had a higher regression coefficient than geographic distance, and only phenological distance significantly predicted genetic distance (geographic distance: β = −0.03, P = 0.90; phenological distance: β = 0.29, P = 0.03). The pattern was the same for \(F^{\prime}_{ST}\) (geographic distance: β = 0.03, P = 0.89; phenological distance: β = 0.30, P = 0.02). When difference in elevation among sites was included as an independent variable in addition to geographic and phenological distances, no independent variable significantly predicted F ST (geographic distance: β = −0.02, P = 0.92; elevation: β = 0.14, P = 0.38; phenological distance: β = 0.25, P = 0.11). However, with elevation included as an independent variable phenological distance did significantly predict \(F^{\prime}_{ST}\) (geographic distance: β = −0.03, P = 0.89; elevation: β = 0.01, P = 0.96; phenological distance: β = 0.30, P = 0.05).

Relationship between genetic distance \(F^{\prime}_{ST}\) and Phenological distance (the number of days apart plants at two sites began flowering). The corresponding Mantel test revealed a significant association of genetic distance and phenological distance
Discussion
We found significant genetic structuring and an association of flowering time and genetic differentiation in the monocarpic montane perennial I. aggregata. Many studies that attempt to explain patterns of genetic structure incorporate distance or landscape factors (Manel and Holderegger 2013). Adding phenological data may generally help better explain the geographic distribution of genetic variation (Hendry and Day 2005). This is particularly important in systems in which interacting species may influence levels of gene flow in populations of their partner species. For plants, gene flow occurs via pollen transfer among co-flowering plants, and phenological differences among plants can contribute to genetic distances among them (Hirao and Kudo 2004; Yamagashi et al. 2005; Hirao and Kudo 2008). In this study, we incorporated flowering time into statistical frameworks used in landscape genetics analyses, and we found that flowering time, but not distance or elevation, was significantly correlated with genetic structure of I. aggregata, both within a meadow over 80 m and over a large geographic area within which the farthest two sampling locations were 130 km.
We examined the association between phenological distance and genetic distance using both Mantel tests and the MMRR method described in Wang (2013). Mantel tests have been the standard way to test for an association among distance matrices in landscape genetic studies, but their accuracy has recently been questioned (Raufaste and Rousset 2001; Legendre and Fortin 2010; Miermans 2015). In addition to more appropriate Type I error rates, an advantage of MMRR over Mantel tests is that it allows variables to be ranked in terms of their effects on the dependent variable. This is useful in cases where one variable may contribute less than others to the dependent variable (in our case genetic distance) but still have a significant effect. The results of MMRR were largely consistent with our Mantel and partial Mantel test results. When geographic and phenological distances were used to predict genetic distance, phenological distance, but not geographic distance, was significantly associated with genetic distance, which was also apparent from the Mantel results. When elevation was included in the model used to predict genetic distance, model coefficients consistently showed the strongest relative effect for phenological distance. None of the variables significantly predicted F ST but phenological distance did significantly predict \(F^{\prime}_{ST}\). This was somewhat consistent with the Mantel results, in which \(F^{\prime}_{ST}\) was more strongly correlated with phenological distance than F ST , but in which phenological distance was significantly associated with both F ST and \(F^{\prime}_{ST}\) when controlling for elevation. The use of multiple regression in landscape genetics is still in its infancy, and additional studies will be valuable to evaluate its accuracy. Nevertheless, a general consistency between statistical approaches suggests our inferences regarding the effect on phenological on genetic distances are robust.
Why was flowering time strongly associated with patterns of genetic variation? First, genetic control over flowering time could result in genetically similar plants flowering at similar times within meadows. Reduced gene flow between plants that vary in alleles governing flowering time could then lead to differences at nuclear microsatellite loci. A genetic component to flowering time has been found in a range of plant species (Jung and Müller 2009), including I. aggregata, in which the narrow sense heritability of date of first flowering was estimated to be 0.29 (Junger and Bergelson 2000). Second, flowering time in I. aggregata is influenced by microclimatic conditions that affect the timing of snowmelt (Price and Waser 1998). Temporal (year-to-year) consistency in flowering time could result in enhanced genetic differentiation due to genetic drift among groups of co-flowering plants. Indeed, previous studies found associations between genetic differentiation and the timing of snowmelt (Hirao and Kudo 2004; Yamagashi et al. 2005; Hirao and Kudo 2008; Shimono et al. 2009) or elevation (Mitton et al. 1977). We found a positive association between flowering time of I. aggregata and elevation, so it is plausible that flowering time among geographic locations may be consistent over years. However, we found no statistical relationship between genetic differentiation and elevation, which suggests that elevation is not necessarily a good proxy for flowering time of I. aggregata, possibly due to the importance of other factors—such as aspect and soil composition—in determining flowering time. Third, an interesting possible explanation for the observed association between flowering time and genetic distance is that some of the plants sampled were descendants from hybridization events between I. aggregata and a closely related species, I. tenuituba, which is found near the geographic area sampled, often at higher elevations. It is unlikely that any sampled plants were first generation hybrids because I. tenuituba has white, narrow, elongated corollas, hybrids have bright pink, slightly shorter corollas (Meléndez-Ackerman et al. 1997) and we sampled only in areas in which all plants had bright red corollas. Nevertheless, this is an interesting possibility and would be worth investigating if species-specific markers are developed.
It was surprising that even within a meadow, plants that began flowering more than 1 week apart were significantly genetically differentiated. This is consistent with temporal isolation and early stages of reproductive isolation. Temporal isolation within populations has been observed with other species of plants (Antonovics 2006) and animals (e.g. Pacific salmon; Kovach et al. 2013). Temporal isolation of this type may be unlikely in Ipomopsis aggregata because plants can sometimes produce tens of flowers that open successively over time, and within our focal meadow there was some overlap in flowering time between early and later flowering plants (S. Suni, personal observation). An alternative explanation is that separate cohorts may flower at different times. Campbell (1997) found that the age at which I. aggregata plants flower ranges from 2 to 10 years, so it is possible that the genetic differentiation observed could be due to among-cohort genetic drift in cohorts that flower at slightly different times during the summer. Additionally, though early and later flowering plants were intermixed geographically we cannot rule out the possibility that flowering time differences were driven by microenvironmental differences that persist over time.
The observed structure among sites appears to be weak but biologically meaningful. The fact that inference of K > 1 occurred only when we used a location prior with STRUCTURE is consistent with weak divergence among populations. However, low values of the r parameter supported inference of a correlation between geographic location and genetic differentiation. Care must also be used in interpreting our STRUCTURE results because evidence for elevated inbreeding within populations of this species violates an assumption of the model. Mild departures from the model used to partition individuals into genetic groups can lead to an overestimation of the number of clusters with STRUCTURE (Falush et al. 2003). In particular, if linkage disequilibrium among loci is due not to actual population structure, but instead to inbreeding, K might be overestimated. In our case, the third genetic group had only one Hardy–Weinberg violation and one significant LD test, suggesting that it is not inbreeding that resulted in it seeming genetically distinct. For the remaining sites, there was no significant correlation of number of departures from Hardy–Weinberg equilibrium or F IS and the proportion of the genome assigned to the second genetic group among sites. However when the site with the lowest F IS was removed from the analysis, F IS was significantly correlated with the proportion of the genome assigned to the second genetic group, suggesting that inbreeding could be partially responsible for STRUCTURE’s delineations.
The results from STRUCTURE were supported by analyses based on F-statistics and PCA. These results suggest that gene flow occurs within the central drainage and that genetic drift might be responsible for differentiation of the sites that are more peripheral to this drainage. Gene flow within the central drainage could occur via pollinator sharing among co-flowering plants. Gene flow in I. aggregata is thought to occur primarily by pollen transfer by broad-tailed and rufous hummingbirds (Selasphorus platycercus and S. rufus), but bumble bees such as Bombus appositus, solitary bees, and butterflies also visit (Waser 1982). In years when bumble bees are frequent visitors they are likely important pollinators because they transfer on average three times as much pollen and elicit about four times as much seed production as hummingbirds (Mayfield et al. 2001). However, it is unknown how far insects transfer pollen, or how frequently insects visited the I. aggregata plants that were the parents of those sampled in this study. Previous work using dyes as pollen analogues showed that rufous hummingbirds travel on average 0.8 m between plants when foraging within a meadow (Waser 1982), and that more than 95 % of transfer of an experimental pollen analog seemed to occur within 5 m (Waser and Price 1983). However, both species of hummingbirds presumably could transfer pollen longer distances. Alternatively, seed dispersal could occur via water transport. Previous work estimated that the root mean square seed dispersal distance was 0.6 m (Waser and Price 1983), though it is conceivable that during heavy rains, seeds may be washed down slopes. Evaluating the extent to which seed dispersal contributes to gene flow in I. aggregata would require additional tests.
For the peripheral sites, allele frequencies at the relatively few loci we examined might have drifted in the same direction by chance. However, if genetic drift was responsible for the observed pattern we may expect a corresponding decrease in heterozygosity and allelic richness within the peripheral sites, but no such pattern was found. Another possibility is that the breeding structure of I. aggregata influenced the way in which individuals were partitioned into genetic groups. Ipomopsis aggregata exist as hardy rosettes for a number of years before bolting, flowering, and senescence. If different sites were made up largely of plants that germinated in different years, genetic differences among cohorts could be at least partially responsible for the modest allele frequency divergence we observed. The observed higher mean F IS in peripheral sites suggests that inbreeding is also at least partially responsible for our STRUCTURE results. Even if the STRUCTURE results were partially driven by inbreeding as a violation of the underlying model, these results remain important from a conservation perspective. Small peripheral sites with elevated inbreeding levels could be suffering from inbreeding depression and may have lower resilience.
Our results are consistent with those obtained from previous population genetic studies of I. aggregata. We found significant genetic differentiation among sites that were as little as 2.6 km apart, and significant differentiation within a site among plants with different flowering phenology. Previous population genetic analyses using allozymes revealed positive spatial autocorrelation of allele frequencies over just 5 m (Campbell and Dooley 1992), and significant structuring over 25 km (Wolf and Campbell 1995). Interestingly, Wolf and Campbell (1995) found that genetic structure increased more from 250 m to 2.5 km than it did from 2.5 to 25 km, which was likely the result of family structure.
Our results also suggest that strong family structure occurs within sites; we found high F IS values and heterozygote deficits within some sites, which can occur when there is breeding among close relatives (Wright 1922). This may seem surprising given the mating system of I. aggregata, in which plants are self-incompatible and may be expected to mate disassortatively with unrelated individuals. However, It is not uncommon for self-incompatible plants to show a deficiency of heterozygotes (Levin 1978; Turner et al. 1982; Barluenga et al. 2011), and four pieces of evidence suggest that a deficit of heterozygotes may actually be expected in populations of I. aggregata. First, the combination of limited seed dispersal and a propensity of pollinators to move between neighboring plants (Waser 1982) means that plants may often mate with close relatives. Second, I. aggregata seems to suffer from outbreeding depression over just 100 m (Waser and Price 1989), suggesting that to some extent matings between individuals with more similar genotypes may be more successful relative to those with less similar genotypes. Third, many of the sites with higher FIS values were in disturbed areas along trail or roadsides, and, thus may not be part of large, stable populations. These possibly more ephemeral populations seem to have relatively high inbreeding rates, which is likely partially responsible for the FIS observations. It is possible that higher inbreeding rates lead to inbreeding depression (reduced fitness associated with inbreeding) in small populations that occur in suboptimal habitat. Evidence that inbreeding depression occurs in small I. aggregata populations comes from a study that found larger fitness gains in small relative to large populations following pollen transfer (Heschel and Paige 1995). Finally, a deficiency of heterozygotes relative to Hardy–Weinberg expectations could result from sampling individuals that belong to multiple randomly mating cohorts. The I. aggregata that we sampled could have germinated in different years, which could also contribute to the observed deficiency of heterozygotes.
Overall, our results suggest that patterns of genetic diversity and differentiation in I. aggregata may be partially explained by inbreeding within sites and phenological differences within and among sites. Sites in disturbed, human dominated areas along trail or roadsides had lower genetic diversity and higher F IS than sites in more natural meadows, suggesting a possible positive association between anthropogenic disturbance and localized population extinctions and a negative association between anthropogenic disturbance and adaptive potential for I. aggregata. However, the markers used to assess these patterns were neutrally evolving microsatellites, so it would be useful to investigate the extent to which genome-wide patterns of genetic diversity and local adaptation differ among plants in disturbed versus more natural areas. In terms of structure among sites, cohesion we observed among sites in the main drainage may have been the result of pollinator movement, and pollinators may have been cueing in on landscape factors not included in our analysis, such as lack of tree cover, when travelling. Alternatively, rain-mediated seed dispersal down drainages may partly contribute to gene flow, although seed dispersal may be expected to decrease the association of flowering time and genetic structure. The strong association between flowering time and genetic structure that we observed suggests that pollinator-mediated pollen movement among co flowering plants is an important mechanism by which gene flow occurs in I. aggregata. These results have important implications for understanding how the geographic distribution of genetic variation might change under future climate change scenarios that likely include phenological changes such as flowering time.
References
Allendorf FW, Luikart G (2007) Genetics and the conservation of populations. Blackwell Publishing, Hoboken
Antonovics J (2006) Evolution in closely adjacent plant populations X: long-term persistence of prereproductive isolation at a mine boundary. Heredity 97:33–37
Barluenga M, Austerliz F, Elzinga JA, Teixeira S, Goudet J, Bernasconi G (2011) Fine-scale spatial genetic structure and gene dispersal in Silene latifolia. Heredity 106:13–24
Benjamini Y, Yukutieli D (2001) The control of the false discovery rate in multiple testing under dependency. Ann Stat 29:1165–1188
Campbell DR (1997) Genetic and environmental variation in life history traits of a monocarpic perennial: a decade-long field experiment. Evolution 51:373–382
Campbell DR, Dooley JL (1992) The spatial scale of genetic differentiation in a hummingbird-pollinated plant: comparison with models of isolation by distance. Am Nat 139:735–748
Colautti RI, Barrett SCH (2013) Rapid adaptation to climate facilitates range expansion of an invasive plant. Science 342:364–366
Cortés AJ, Waeber S, Lexer C, Sedlacek J, Wheeler JA, van Kleunen M et al (2014) Small-scale patterns in snowmelt timing affect gene flow and the distribution of genetic diversity in the alpine dwarf shrub Salix herbacea. Heredity. doi:10.1038/hdy.2014.19
Dunne JA, Harte J, Taylor KJ (2003) Subalpine meadow flowering phenology responses to climate change: integrating experimental and gradient methods. Ecol Monogr 73:69–86
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Cons Gen Res 4:259–361
Ennos RA (1994) Estimating the relative rates of pollen and seed migration among plant populations. Nature 72:250–259
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Falush D, Stephens M, Pritchard J (2003) Inference of population structure using multilocus genotypic data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Fenster CB (1991) Gene flow in Chamaecrista fasciculate (Leguminosae) I. Gene dispersal. Evolution 45:398–409
Fox GA (2003) Assortative mating and plant phenology: evolutionary and practical consequences. Evolut Ecol Res 5:1–18
Frankham R (2010) Challenges and opportunities of genetic approaches to biological conservation. Biol Conserv 143:1919–1927
Freeman RS, Brody AK, Neefus CD (2003) Flowering phenology and compensation for herbivory in Ipomopsis aggregata. Oecologia 136:394–401
Geber MA, Griffen LR (2003) Inheritance and natural selection on functional traits. Int J Plant Sci 164:S21–S42
Gerber J-D, Baltisberger M, Leuchtmann A (2004) Effects of a snowmelt gradient on the population structure of Ranunculus alpestris. Bot Helv 114:67–78
Gilpin M, Hanski I (1991) Metapopulation dynamics: empirical and theoretical investigations. Academic Press, London
Hall MC, Willis JH (2006) Divergent selection on flowering time contributes to local adaptation in Mimulus guttatus populations. Evolution 60:2466–2477
Hendry AP, Day T (2005) Population structure attributable to reproductive time: isolation by time and adaptation to time. Mol Ecol 14:901–916
Heschel MS, Paige KN (1995) Inbreeding depression, environmental stress, and population size variation in Scarlet Gilia (Ipomopsis aggregata). Conserv Biol 9:126–133
Hirao AS, Kudo G (2004) Landscape genetics of alpine-snowbed plants: comparisons along geographic and snowmelt gradients. Heredity 93:290–298
Hirao AS, Kudo G (2008) The effect of segregation of flowering time on fine-scale spatial genetic structure in an alpine-snowbed herb Primula cuneifolia. Heredity 100:424–430
Holderegger R, Wagner HH (2008) Landscape genetics. Bioscience 58:199–207
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Mol Ecol Res 9:1322–1332
Inouye DW, McGuire AD (1991) Effects of snowpack on timing and abundance of flowering in Delphinium nelsonii (Ranunculaceae): implications for climate change. Am J Bot 78:997–1001
Jay F, Manel S, Alvarez N, Durand EY, Thuiller W, Holderegger R, Taberlet P, François O (2012) Forecasting changes in population genetic structure of alpine plants in response to global warming. Mol Ecol 21:2354–2368
Jost L (2008) GST and its relatives do not measure differentiation. Mol Ecol 17:4015–4026
Jung C, Müller AE (2009) Flowering time control and applications in plant breeding. Trends Plant Sci 14:563–567
Junger T, Bergelson Y (2000) The evolution of compensation to herbivory in Scarlet Gilia, Ipomopsis aggregata: herbivore-imposed natural selection and the quantitative genetics of tolerance. Evolution 54:764–777
Keller SR, Levsen N, Ingvarsson PK, Olson MS, Tiffin P (2011) Local selection across a latitudinal gradient shapes nucleotide diversity in balsam poplar, Populus balsamifera L. Genetics 188:941–952
Kittelson PM, Maron JL (2001) Fine-scale genetically based differentiation of life history traits in the perennial shrub Lupinus arboreus. Evolution 55:2429–2438
Korves TM, Schmid KJ, Caicedo AL, Mays C, Stinchcombe JR, Purugganan MD, Schmitt J (2007) Fitness effects associated with the major flowering time gene FRIGIDA in Arabidopsis thaliana in the field. Am Nat 169:E141–E157
Kovach RP, Gharrett J, Tallmon DA (2013) Temporal patterns of genetic variation in a salmon population undergoing rapid change in migration timing. Evol App 6:795–807
Lambert AM, Miller-Rushing AJ, Inouye D (2010) Changes in snowmelt date and summer precipitation affect the flowering phenology of Erythronium grandiflorum (glacier lily; Liliaceae). Am J Bot 97:1431–1437
Legendre P, Fortin M (2010) Comparison of the Mantel test and alternative approaches for detecting complex multivariate relationships in the spatial analysis of genetic data. Mol Ecol Res 10:831–844
Levin D, Kerster HW (1974) Gene flow in seed plants. Evol Biol 7:139–220
Levin D (1978) Genetic variation in annual Phlox: self-compatible versus self-incompatible species. Evolution 32:245–263
Manel S, Holderegger R (2013) Ten years of landscape genetics. Trends Ecol Evol 28:613–621
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197
Mayfield MM, Waser NM, Price MV (2001) Exploring the ‘most effective pollinator principle’ with complex flowers: bumblebees and Ipomopsis aggregata. Ann Bot-Lond 88:591–596
Meirmans PG, Van Tienderen PH (2004) GENOTYPE and GENODIVE: two programs for the analysis of genetic diversity of asexual organisms. Mol Ecol Notes 4:792–794
Meléndez-Ackerman E, Campbell DR, Waser NM (1997) Hummingbird behavior and mechanisms of flower color in Ipomopsis. Ecology 78:2532–2541
Miermans PG (2015) Seven common mistakes in population genetics and how to avoid them. Mol Ecol 13:3223–3231
Miermans PG, Hedrick PW (2011) Assessing population structure: F ST and related measures. Mol Ecol Res 11:5–18
Mitton JB, Linhart YB, Hamrick JL, Beckman J (1977) Population differentiation and mating systems in ponderosa pine in the Colorado Front range. Theor App Genet 51:5–14
Narum SR (2006) Beyond Bonferroni: less conservative analyses for conservation genetics. Con Gen 7:783–787
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Oksanen JF, Guillaume B, Kindt R, Legrende R, et al (2011) Vegan: community ecology package. R package version 1.17-10. http://cran.r-project.org/web/packages/vegan/
Parmesan C, Yohe G (2003) A globally coherent fingerprint of climate change impacts across natural systems. Nature 421:37–42
Peakall ROD, Smouse PE (2006) genalex 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:228–295
Price M, Waser N (1998) Effects of experimental warming on plant reproductive phenology in a subalpine meadow. Ecology 79:1261–1271
Pritchard JM, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Core Team (2008) A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
Raufaste N, Rousset F (2001) Are partial Mantel tests adequate? Evolution 55:1703–1705
Rousset F (2008) GENEPOP’007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Res 8:103–106
Ryman N, Palm S, André C, Carvalho GR, Dahlgren TG, Jorde PE, Laikre L, Larsson LC, Palmé A, Ruzzante DE (2006) Power for detecting genetic divergence: differences between statistical methods and marker loci. Mol Ecol 15:2031–2045
Saccheri I, Kuussaari M, Kankare M, Vikman P, Fortelius W, Hanski I (1998) Inbreeding and extinction in a butterfly metapopulation. Nature 392:491–494
Shimono Y, Watanabe M, Hirao AS, Wada N, Gaku K (2009) Morphological and genetic variations of Potentilla matsumurae (Rosacae) between fellfield and snowbed populations. Am J Bot 96:728–737
Shindler et al (2010) Population diversity and the portfolio effect in an exploited species. Nature 465:609–613
Soulé ME (1987) Viable populations for conservation. Cambridge University Press, Cambridge
Stanton ML, Galen C, Shore JS (1997) Population structure along a steep environmental gradient: consequences of flowering time and habitat variation in the snow buttercup, Ranunculus adoneus. Evolution 51:79–94
Stanton ML, Roy BA, Thiede DA (2000) Evolution in stressful environments. I. Phenotypic variability, phenotypic selection, and response to selection in five distinct environmental stresses. Evolution 54:93–111
Stinchcomb JR, Weinig C, Ungerer M, Olsen KM, Mays C, Halldorsdottir SS, Purugganan MD, Schmitt J (2004) A latitudinal cline in flowering time in Arabidopsis thaliana modulated by the flowering time gene FRIGIDA. Proc Nat Acad Sci 101:4712–4717
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits L (2007) Putting the “landscape” in landscape genetics. Heredity 98:128–142
Turner ME, Stephens JC, Anderson WW (1982) Homozygosity and patch structure in plant populations as a result of nearest-neighbor pollination. Proc Nat Acad Sci 79:203–207
Wang JI (2013) Examining the full effects of landscape heterogeneity on spatial genetic variation: a multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution 67:3403–3411
Waser NM (1982) A comparison of distances flown by different visitors to flowers of the same species. Oecologia 55:251–257
Waser NM, Price MV (1983) Optimal and actual outcrossing, and the nature of plant-pollinator interaction. In: Jones CE, Little RJ (eds) Handbook of experimental pollination biology. Van Nostrand Reinhold, New York, pp 341–359
Waser NM, Price MV (1989) Optimal outcrossing in Ipomopsis aggregata: seed set and offspring fitness. Evolution 43:1097–1109
Weis AE, Kossler TM (2004) Genetic variation in flowering time induces phenological assortative mating: quantative genetic methods applied to Brassica rapa. Am J Bot 91:825–836
Wolf PG, Campbell DR (1995) Hierarchical analysis of allozymic and morphometric variation in a montane herb, Ipomopsis aggregata (Polemoniaceae). J Heredity 86:386–394
Wright S (1922) Coefficients of inbreeding and relationship. Am Nat 56:330–338
Wright S (1943) Isolation by distance. Genetics 28:114–138
Wright S (1951) The genetical structure of natural populations. Ann Eug 15:323–354
Wu C (2006) Characterization of microsatellite loci in Ipomopsis (Polemoniaceae) wildflowers. Mol Ecol Res 6:921–923
Yamagashi H, Allison TD, Ohara M (2005) Effect of snowmelt timing on the genetic structure of an Erythronium grandiflorum population in an alpine environment. Ecol Res 20:199–204
Acknowledgments
We thank Matt Kaplan and the staff of the Genomics Core Facility at the University of Arizona for help with microsatellite genotyping; the Rocky Mountain Biological Laboratory in Gothic, CO, for support and use of facilities and equipment, and Judith Bronstein and Nickolas Waser for help and guidance during fieldwork. This work was funded in part also by NIH Grant 5 K12 GM000708, which was awarded to the Center for Insect Science at the University of Arizona, and by support from the University of Massachusetts Amherst to S. Suni while she was the Darwin Fellow.
Author information
Authors and Affiliations
Corresponding author
Additional information
Data accessibility
Microsatellite genotypes will be submitted to Dryad Digital Repository for archiving.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Suni, S.S., Whiteley, A.R. Genetic structure of a montane perennial plant: the influence of landscape and flowering phenology. Conserv Genet 16, 1431–1442 (2015). https://doi.org/10.1007/s10592-015-0751-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-015-0751-z