
Abstract
Cattle (Bos taurus) and goat (Capra hircus) belong to the Bovidae family, and they share a common ancestor 19.7–21.5 Ma ago (MYA). The Bovidae family apparently experienced a rapid species radiation in the middle Miocene. The present day cattle and goat possess the same diploid chromosome number (2n = 60) and structurally similar autosomes, except that a small subcentromeric portion of cattle chromosome nine has been translocated to goat chromosome 14. In this study, we adopted a new strategy that involves the use of bioinformatics approach to detect unknown cryptic chromosome divergences between cattle and goat using and subsequent validation using the fluorescence in situ hybridization (FISH) of bacterial artificial chromosome clones. We identified two hypothetical discrepancies between the cattle and goat genome assemblies: an inversion in the goat chromosome 13 and a transposition in the goat chromosome 6. The FISH technique allowed clear validation of the existence of a new 7.4 Mb chromosomal inversion in the goat chromosome 13. Regarding the transposition in the goat chromosome six, FISH analyses revealed that the cattle and goat genomes shared the same organization, with the assembly of the goat genome being the correct one. Moreover, we defined, for the first time, the size and orientation of the translocated fragment involved in the evolutionary translocation between cattle chromosomes 9 and goat chromosome 14. Our results suggest that bioinformatics represents an efficient method for detecting cryptic chromosome divergences among species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Both cattle (Bos taurus) and goat (Capra hircus) belong to family Bovidae, order Cetartiodactyla, suborder Ruminantia. The Bovidae family can be divided in two subfamilies: Bovinae and Antilopinae. Bovinae includes three tribes: Bovini, Tragelaphini, and Boselaphini, while Antilopinae includes nine tribes: Aepycerotini, Neotragini, Antilopini, Reducini, Oreotragini, Cephalophini, Alcelaphini, Hippotragini, and Caprini (Hassanin et al. 2012). Cattle belong to the Bovini tribe, whereas goat belongs to the Caprini tribe (Fig. 1).

Evolution tree of Bovidae family. The figure reports the position of the 12 tribes present in the Bovidae family in a evolutionary tree (adapted from Hassanin et al. 2012). The distances between tribes are not representative of the evolution time
The common ancestor belonging to Cetartiodactyla has been dated back to 60–77 Ma ago (MYA) (Arnason and Gullberg 1996; Bininda-Emonds et al. 2007), while the first presence of Bovidae has been set at 23 MYA (Vrba 1979; Kingdom 1989). The closest ancestor of cattle and goat has been traced back to the phylogenetic separation between Bovinae and Antilopinae, which is dated in the early Miocene between 19.7 and 21.5 MYA (Hassanin et al. 2012). After this separation, a rapid speciation involving the family Bovidae probably occurred in the middle Miocene. Currently, the Bovidae family consists of more than 149 species, including the most important zoo-economic species such as cattle, sheep, goats, buffalo, and zebu. From the chromosomal perspective, all bovid species possess similar fundamental chromosome numbers (58–62), but highly variable diploid numbers (30–60). This difference may be largely due to the high incidence of autosomal centric fusions that occurred during the evolutionary process of the Bovidae family (Iannuzzi et al. 2009). Cattle and goat possess the same diploid number (2n = 60) and structurally similar autosomes; to date, only one small karyotypic difference has been detected, that is, a small translocation, with the subcentromeric portion of cattle chromosome 9 (BTA9) being translocated to the goat chromosome 14 (CHI14) (ISCNDB 2000). This rearrangement was first demonstrated by linkage analysis (de Gortari et al. 1998) and later confirmed by FISH-mapping (Iannuzzi et al. 2001). This translocation could represent the leading ramification of Bovinae and Antilopinae divergence (Buckland and Evans 1978). However, to date no information is available regarding the size of the translocated fragment.
In this study, we report a new strategy that allows us to detect cryptic chromosome divergences between cattle and goat using bioinformatic and FISH-mapping approaches.
Materials and methods
Bioinformatics
Efficient E-probes were produced for comparing the cattle and goat genomes using the bacterial artificial chromosome (BAC) END sequences (ENDs) as probes because (a) we believed that the two ENDs of a single BAC (referred to as TJ and TV) belong to the same DNA sequence as chimeric cloning is extremely rare in the BACs library construction; (b) in a well-assembled genome around the BAC region, the two ENDs of a single BAC must be oriented in opposite direction; and (c) the two ENDs must be at a distance >30 and <300 Kb as this size represents the most common size of an insert in the BAC library. We downloaded all of the ENDs belonging to the INRA Cattle BAC Library from the GSS database (NCBI), retaining only those BACs that showed both ENDs (discarding the single END). The analysis was performed with 24,743 BACs and the corresponding 49,486 ENDs.
To locate the sequence of each BAC in the cattle and goat genomes, we performed a local alignment of each BAC-END on the whole cattle genome and on the whole goat genome by using BLAT (Kent 2002). To accelerate the process, we used a local client/server version of BLAT. When BLAT is used to search for a particular sequence in a genome, the first step before the alignment is the indexation of the genome. Indexation is an internal strategy of the algorithm, an automatic process that is completed within few seconds. There are two ways of aligning few sequences: using a web interface or by installing a local version of BLAT in a computer. However, if several thousands of sequences are to be aligned, this process would be critically compromised by time consumption as the genome is indexed before each query or alignment. Building an indexed genome database and maintaining it in memory throughout the entire process is possible by using a client/server version of BLAT that is installed in the local server. Thus, the genome is indexed only once at the beginning, following which each query can access the server by passing the need to index the genome for each query. The options used for the alignments were, “tileSize = 11, minMatch = 4, stepSize = 5, repMatch = 2253”; these values are similar to the default values suggested by BLAT developers for aligning the DNA sequences, albeit with slight modification to increase the stringency conditions of the alignment. Often, each query yields several alignments of the query sequence with different regions in the reference genome. We retained only those alignments that have 99.5 % or more identity. The output file for each alignment was further processed to investigate whether the two ENDs of each BAC fulfilled the requirement of being in the opposite orientation in the same chromosome and at a distance of 30–300 Kb. The bioinformatic pipeline is depicted in Fig. 2.

Bioinformatics pipeline. The figure summarizes the passage performed in order to identify the useful BACs. The data contained in the blue box were obtained from GSS database (NCBI). The green box identifies the operations performed against the cattle genome assembly. The red box identifies the operations performed against the goat genome assembly
Fluorescent in situ hybridization (FISH)
FISH experiments were performed according to the protocol described by De Lorenzi et al. (2014). The BACs used as probes belonged to the INRA Library (Eggen et al. 2001) and are reported in Table 1.
Genome version
To identify the position of the BACs, we used the following freely available genome assembly: bosTau6 for cattle (http://genome.ucsc.edu; Zimin et al. 2009) and genome browser v1.0 for goat (http://goat.kiz.ac.cn/GGD/index; Dong et al. 2013).
Statistical analysis
Data regarding the average distance (Kb) between consecutive BACs were statistically analyzed using the GLM procedure of the SAS package (2008).
Results
BACs localization on the cattle genome
Initially 24,743 BACs were subject to the in silico analysis. Twenty-five BACs were discarded as both ENDs of each of these 25 clones showed no chromosomal localization in cattle; another 2330 BACs were also discarded as they each showed ENDs localization on two different cattle chromosomes. For reliable E-probes, each END must have only one genomic localization; we thus discarded a further 2174 BACs as they showed more than one “strong” genomic localizations for at least one END. After applying the coverage filter (i.e., % of END bps that match with the identified genome region) of the ENDs, 1805 BACs were discarded as their ENDs showed coverage of <190 % (each END can show a maximum coverage of 100 %). The remaining BACs were further filtered according to their similitude (i.e., % of END bps identical to the genome region). Only those BACs whose ENDs had a combined value of similitude >198 % (max possible = 200 %) were maintained, this process led to 1158 BACs being discarded. Finally, a total of 266 BACs with an insert size (measured as the distance among the ENDs localized on the genome) of <30 Kb, 110 BACs with an insert size >301 Kb, and 285 BACs with ENDS not oriented in opposite orientation were discarded. After the abovementioned filtering procedures were completed, we obtained a total of 16,589 BACs with extremely reliable localizations on the cattle genome. In order to generate BACs representative of all cattle genomes, we observed their distribution on each chromosome. Table 2 depicts the BAC distribution data on single chromosomes. Considering the whole genome, we had a BAC every 160 Kb, and almost all of the chromosomes showed comparable density. However, the distribution of the BACs on individual chromosomes does not appear to be statistically uniform. BTAX and BTA15 had greater BAC distance average, whereas BTA25 and BTA26 had lesser distance average. The results of the statistical analysis are shown in Table 2.
BACs localization on goat genome
In order to obtain a reliable localization of the BACs on goat genome, we conducted an analysis and a subsequent check control as reported earlier for cattle. We discarded 3583 BACs because their ENDs were localized on two different chromosomes as well as 184 BACs as they possessed at least one END with more than one localization on the goat genome. Finally, we discarded 3 BACs with a “goat” insert size of <30 Kb as well as 108 BACs with an insert size of >301 Kb. At the end of this process, we obtained 12,711 BACs, each with a reliable localization on both the cattle and goat genomes, which allowed a good comparison between the two genomes.
Visualization of the differences
Cattle and goat showed similar karyotype (excluding the sexual chromosomes), with the only difference being the BTA9;14 small translocation. To reveal the potential cryptic differences present between the two genomes, for each single chromosome, we represented the position of each BAC using a Cartesian graph (Dong et al. 2013). When all BACs were located in an analogous position within the same chromosome, a straight line appeared (Fig. 3a). Following this procedure, we identified three hypothetical autosomal discrepancies between the two genomes, including the previously reported BTA9;14 translocation (Fig. 3b); an inversion detected in chromosome 13 of goat (CHI13-, Fig. 3c), and a transposition in goat chromosome 6 (CHI6-, Fig. 3d). As expected, more complex structural differences were also detected between the cattle and goat X chromosomes assembly. However, the description and validation of such complex rearrangements were beyond the scope of the current paper.

Output of the bioinformatic analysis. The figure reports the graphic output of the comparison among BACs localization on cattle and goat genomes. a Each dot represents a BAC. In the case of a perfect correlation between cattle (x axis) and goat (y axis) position of BACs a straight line appears. b Visualization of the BTA9;14 translocation in goat; the red arrow shows that cattle genome portion is translocated on CHI14. The orientation of the line in this portion, inverted compared to the remaining part, indicates that the fragment is also inverted. c Visualization of the inversion of CHI13. The green arrow indicates the portion of the cattle genome that appears to be inverted in goat genome. d Visualization of the transposition of CHI6. The black arrow indicates the portion of the cattle genome that seems to be transposed at the end of chromosome 6 of goat
BTA9;14 translocation
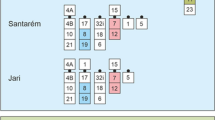
From the information provided by the localization of the BACs in the centromeric region of BTA9, we assumed that the translocated portion was also inverted in the goat chromosome 14 and also that this region was 13-Mb long (Fig. 3b; Table 3). The use of FISH technology demonstrated that both assumptions were correct. In particular, using the BAC 307H04 and 636E01, we verified that the translocated portion of BTA9 was also inverted in the goat genome. In cattle, FISH experiments were performed on well-spread metaphases belonging to a rcp9;11 carrier to identify the BTA9 (De Lorenzi et al. 2007); in goat, the CHI14 was identified along with the BAC 456B03 as a marker (Fig. 4a, b). Then, using BAC 513C11, we highlighted that the length of the translocated fragment was 12.8 and 13.2 Mb (Fig. 4c, d). Goat chromosome 9 was identified using BAC 474A12 as a marker.

FISH analyses. The figure reports the results obtained after FISH experiments performed to confirm or denied the bioinformatic outputs. In all images the name of the BACs corresponds to FISH signals. a, c, e, g FISH on cattle. b, d, f, h FISH on goat. a–d FISH to analyzes the BTA9;14 translocation. e, f FISH to analyze the CHI13 inversion. g, h FISH to analyze the CHI6 transposition
Goat chromosome 13 inversion
Observing the localization of the BACs in the cattle and goat genomes, we could conclude that a 5.3-Mb long region was inverted in the goat genome in comparison with the cattle genome (Fig. 3c; Table 4). We verified this hypothesis by FISH. Using the BACs 918D08 and 212F04 as probes, we demonstrated that the fragment was actually inverted in the goat when compared with the cattle (Fig. 4e, f).
Goat chromosome 6 transposition
Finally, we considered the hypothetical transposition evidenced by the bioinformatic procedure on the goat chromosome 6 (Fig. 3d; Table 5). A short genome fragment of length 3.3 Mb that was believed to be close to the telomere in cattle was found to be positioned in the telomeric position in goat. Using BACs 713F08 and 246G1, we found no differences between the cattle and goat genomes and, surprisingly, we found that the genome assembly reported for goat represents the correct one (Fig. 4g, h).
Discussion
During the BAC mapping on the cattle genome using the bioinformatic approach, we discarded 8154 BACs that represented 33 % of the available total number of BACs (24,743). These BACs did not pass the control checks. Although it is possible that some of these BACs that possessed good localization got eliminated due to the highly restrictive filters we applied. Considering that the cattle genome assembly we used (UMD3.1) contains some gaps (estimated at approximately 8 %; Zimin et al. 2009), some of the discarded BACs probably map to these unknown genomic regions. Some BACs could be discarded as their ENDs were localized in the repetitive regions, in the CNV regions, or on the Y chromosome.
More than 100 BACs were discarded as they showed an insert size of >301 Kb. Some of the BACs showed an insert size of 350–400 Kb that was compatible with the BAC library construction, although others showed an insert size of >1 Mb, a condition that is incompatible with the library construction procedure. Moreover, in the cattle genome, we discarded 285 BACs with ENDs that were not in opposite orientation. In these two last cases, the most probable hypothesis is an incorrect genome assembly in the concerned genomic region due to the use of human synteny in cattle genome assembly. Finally, unexpectedly, the distribution of the BACs on single chromosomes was not statistically similar. The reason for this observation is intriguing, with a plausible explanation that these particular cattle chromosomes probably contain fewer repetitive sequences and, therefore, fewer BACs were discarded at the check-control level.
There were some interesting conclusions for the localization of the 16,589 BACs with good cattle localization on the goat genome. First, 3583 BACs showed good localization of the two ENDs on two different goat chromosomes (in this case, one END coincided with cattle localization). This observation can be explained by the following two hypothetical reasons: either incorrectly assembled goat genome or localization of the BACs at an evolutionary breakpoint between cattle and goat. We believe that the second hypothesis is extremely unlikely considering the high degree of similarity between the cattle and goat genomes as well as the large number of presumed differences between them. Furthermore, this hypothesis is not supported by the strategy that is applied to the construction of the goat genome assembly, whereby the 315 super-scaffolds were constructed based on the physical map generated by optical mapping, but 302 of the 315 super-scaffolds were anchored onto 30 goat pseudo-chromosomes using cattle synteny information obtained from cattle genome assemblies Bta4.0 and UMD3.1 (see Dong et al. 2013 for detail). This means that our bioinformatic approach could identify only such structural differences between cattle and goats that are contained in the super-scaffolds. However, because the super-scaffold was assembled without using any cattle synteny information, the cryptic rearrangements we detected could reflect the true karyotype divergence between the two species if we assume the genome assemblies are correct.
To summarize, we used a bioinformatic approach in this study to obtain a total of 12,711 BACs with a strong localization in the genomes of cattle and goat. We also obtained information that can facilitate improvement of the cattle and goat genomes in the future. Moreover, our strategy enabled us to define, for the first time, the size and orientation of the translocated fragment involved in the BTA9;14 evolutionary rearrangement. Until recently, the inversion of the fragment was only suspected, but not confirmed (Iannuzzi et al. 2001), and its size was unknown. Another interesting discrepancy that we detected was the hypothetical transposition that occurred in the goat chromosome 6. In this case, the FISH analyses revealed that both the cattle and goat genomes shared the same organization, and that the assembly of the goat genome was the correct one. This finding is surprising as the cattle genome is generally believed to be more accurate than the goat genome.
Notably, we identified a new chromosomal divergence between cattle and goat: an inversion of 7.4 Mb in the goat chromosome 13 with respect to its cattle homologue chromosome (BTA14). This discovery represents the starting point for new areas of research, including research on the effect of this inversion on the goat phenotype. The two rearrangement breakpoints (RBP) were included in the regions of 853,611 bp (10,685,991–11,539,602 bp) and 1,260,473 bp (16,805,420–18,065,993 bp) for the centromeric and telomeric RBPs, respectively. The first RBP includes three genes (SNRPB2, OTOR, and NANP), whereas the second one includes 9 genes (PRKCQ, PFKFB3, RBM17, IL2RA, FBXO18, ANKRD26, YME1L1, MASTL, and ACBD5). Further investigations are warranted to identify the precise point where the RBPs occurred, as we cannot exclude that this event interfered with the regulation of one or more of these genes. For example, in pig SCNN1B, a genetic factor involved in the perception of salty taste was located in a porcine-specific evolutionary breakpoint (EBP), and this genomic rearrangement jeopardized the ability to perceive the taste of salt (Groenen et al. 2012). The inverted fragment includes 17 genes (CCDC3, CDC123, NUDT5, SEC61A2, DHTKD1, PROSER2, ECHDC3, USP6NL, CELF2, MIR7861, VAMP7, GATA3, ATP5C1, KIN, ITIH2, ITIH5, and SFMBT2). Further studies are required to understand whether this inversion is responsible for the perturbation of gene expression in goat of one or more of these genes.
Another question that needs to be addressed by future study concerns whether the inversion is present only in goat or whether other Antilopinae tribes also show the same genomic rearrangement. The preliminary bioinformatic result indicates that this inversion is also present in sheep (Ovis aries), although further investigation is needed to highlight its occurrence in other species.
In conclusion, we applied a new bioinformatic strategy to identify the new cryptic rearrangements between cattle and goats and validated their existence in goat, with respect to cattle, using the FISH technique.
Abbreviations
- MYA:
-
Million years ago
- FISH:
-
Fluorescent in situ hybridization
- CHI:
-
Capra hircus
- BTA:
-
Bos taurus
- Mb:
-
Megabase
- BAC:
-
Bacteria artificial chromosome
- INRA:
-
Institut nationale de la recherche agronomique
- Kb:
-
Kilobase
- CNV:
-
Copy number variation
- EBP:
-
Evolutionary break point
References
Arnason U, Gullberg A (1996) Cytochrome b nucleotide sequences and the identification of five primary lineages of extant cetaceans. Mol Biol Evol 13:407–417
Bininda-Emonds OR, Cardillo M, Jones KE et al (2007) The delayed rise of present-day mammals. Nature 446:507–512
Buckland RA, Evans HJ (1978) Cytogenetic aspects of phylogeny in the Bovidae. I. G-banding. Cytogenet Cell Genet 32:64–71
de Gortari MJ, Freking BA, Cuthbertson RP et al (1998) A second-generation linkage map of the sheep genome. Mamm Genome 9:204–209
De Lorenzi L, De Giovanni A, Molteni L, Denis C, Eggen A, Parma P (2007) Characterization of a balanced reciprocal translocation, rcp(9;11)(q27;q11) in cattle. Cytogenet Genome Res 119:231–234
De Lorenzi L, Rossi E, Gimelli S, Parma P (2014) De novo reciprocal translocation t(5;6)(q13;q34) in cattle: cytogenetic and molecular characterization. Cytogenet Genome Res 142:95–100
Dong Y, Xie M, Jiang Y et al (2013) Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat Biotechnol 31:135–141
Eggen A, Gautier M, Billaut A et al (2001) Construction and characterization of a bovine BAC library with four genome-equivalent coverage. Genet Sel Evol 33:543–548
Groenen MA, Archibald AL, Uenishi H et al (2012) Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491:393–398
Hassanin A, An J, Ropiquet A, Nguyen TT, Couloux A (2012) Combining múltiple autosomal introns for studying shallow phylogeny and taxonomy of Laurasiatherian mammals: application to the tribe Bovini (Cetartiodactyla, Bovidae). Mol Phylogenet Evol 66:766–775
Iannuzzi L, Di Meo GP, Perucatti A, Schibler L, Incarnato D, Cribiu EP (2001) Comparative FISH-mapping in river buffalo and sheep chromosomes: assignment of forty autosomal type I loci from 16 human chromosomes. Cytogenet Cell Genet 94:43–48
Iannuzzi L, King WA, Di Berardino D (2009) Chromosome evolution in domestic bovids as revealed by chromosome banding and FISH-mapping techniques. Cytogen Genome Res 126:49–62
ISCNDB 2000 (2001) International system for chromosome nomenclature of domestic bovids. Cytogenet Cell Genet 92:283–299
Kent WJ (2002) BLAT—the BLAST-like alignment tool. Genome Res 12:656–664
Kingdom J (1989) Bovids, horned ungulates. Pp. 1–746 in East African mammals: an atlas of evolution in Africa. Vol. IIIC and D. University of Chicago Press, Chicago
SAS (2008) Release 9.2, SAS Inst. Inc., Cary, N.C., USA
Vrba ES (1979) Phylogenetic analysis and classification of fossil and recent Alcelaphini Mammalia. Bovidae Biol J Linn Soc 11:207–228
Zimin AV, Delcher AL, Florea L et al (2009) A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol 10:R42
Acknowledgments
We are grateful to the animal owner for providing blood samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Fengtang Yang
Rights and permissions
About this article

Cite this article
De Lorenzi, L., Planas, J., Rossi, E. et al. New cryptic karyotypic differences between cattle (Bos taurus) and goat (Capra hircus). Chromosome Res 23, 225–235 (2015). https://doi.org/10.1007/s10577-014-9462-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10577-014-9462-4