
Abstract
Recent studies have shown that exposure to sevoflurane in developing brains causes neuronal apoptosis and cognitive dysfunction. “Necroptosis” is a novel pathway of necrosis. We introduced the caspase-specific inhibitor Z-VAD in addition to the receptor-interacting protein kinase 1 (RIPK1) inhibitor Nec-1, to ascertain the existence and importance of necroptosis. Sprague–Dawley rat pups postnatal day 7 were randomly assigned into one of five groups: control, sevoflurane + Z-VAD, sevoflurane + Nec-1, sevoflurane + Z-VAD + Nec-1 and 3% sevoflurane group. Neuronal apoptosis was evaluated by hematoxylin and eosin staining. The MTT assay was performed to evaluate cell viability. Immunofluorescence was employed to measure expression of RIPK1 and RIPK3. Western blots showing expression of RIPK1, RIPK3 and phosphorylation of mixed lineage kinase domain-like (p-MLKL) were used to explore the role of necroptosis. Binding of RIPK1/RIPK3 was detected via co-immunoprecipitation. Finally, the Morris water maze test was used to determine cognitive function. Exposure to 3% sevoflurane for 6 h induced neurotoxicity and inhibited cell viability. Neuron viability was low in the SEV, SEV + Z-VAD and SEV + Nec-1 groups. The study revealed that RIPK1 and RIPK3 protein expression increased significantly, but there was no significant differences between the SEV and SEV + Z-VAD groups. The expression of p-MLKL significantly increased in the SEV and SEV + Z-VAD groups, but not in the SEV + Nec-1 group or SEV + Z-VAD + Nec-1 group compared to the control group. Co-immunoprecipitation results showed that sevoflurane exposure enhanced binding of RIPK1/RIPK3 protein significantly. Blockade of apoptosis and necroptosis alleviated sevoflurane-induced cognitive impairment. Sevoflurane exposure elicited neurotoxicity within neonatal hippocampal neurons and tissues. Blockade of apoptosis or necroptosis alone did not attenuate sevoflurane-induced neurotoxicity (SIN). RIPK1/RIPK3-mediated necroptosis was involved in SIN in hippocampal neurons. SIN could be attenuated only by inhibiting both apoptosis and necroptosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Anesthesia-induced neurotoxicity in the developing brains of neonates has been illustrated widely in both animals and humans in recent years, and has attracted much attention (Andropoulos and Greene 2017; Davidson and Vutskits 2017). The form of regulated or programmed necrosis is represented by necroptosis, which can be caused by chemical and mechanical injury, inflammation, or infection (Khoury et al. 2020). Necroptosis has emerged as a crucial pathological process involved in many diseases, such as myocardial infarction, stroke, atherosclerosis, ischemia–reperfusion injury, pancreatitis and inflammatory bowel diseases (Linkermann and Green 2014). 14 December 2016, the U.S. Food and Drug Administration issued a drug safety communication warning, stating that if neonates or children less than 3 years of age, (as well as pregnant women in the third trimester) undergo anesthesia for more than 3 h or are repeatedly subjected to anesthetics, brain development may be affected. The warning focused on 11 commonly used general-anesthetic agents that bind to gamma-aminobutyric acid (GABA) or N-methyl-d-aspartate (NMDA) receptors, including sevoflurane, propofol, ketamine, barbiturates, and benzodiazepines.
Sevoflurane is a well-known commonly used inhalation anesthetic. Previous reports suggest that 6 h of exposure to 3% sevoflurane in the developing brain can lead to impairment of synaptic plasticity in the hippocampus, as well as hippocampus-related impairments in learning and memory (Xiao et al. 2016). How sevoflurane affects neuronal development is not completely understood.
Apoptosis (autophagic cell death) and necrosis have been considered the classical pathways of cell death (Van den Berghe et al. 2014). Of these, apoptosis and autophagic cell death are considered the uniquely self-regulated forms of cell death. Early studies identified necrosis as a “passive” pathway of cell death that is often caused by extracellular accidental damage or cytokines, and it is not considered to be regulable (Pasparakis and Vandenabeele 2015; Linkermann and Green 2014).
Necroptosis has been found to be a new pathway of necrosis. It is tightly operated and modulated by genetic controls (de Almagro and Vucic 2015; Huang et al. 2015). Receptor-interacting protein kinase 1 (RIPK1), receptor-interacting protein kinase 3 (RIPK3) and mixed lineage kinase domain-like (MLKL) are all considered important actors in the necroptosis signaling pathway (Vandenabeele et al. 2010; Moriwaki et al. 2015).
Unlike apoptosis, necroptosis is a caspase-independent and programmed form of cell death that can be initiated by activation of death receptors. Apoptosis and necroptosis often coexist, and necroptosis can act as an alternative pathway if apoptosis is suppressed. Apoptosis can be blocked by the pan-caspase inhibitor zVAD-fmk (Z-VAD), and necroptosis can be blocked by the RIPK1 inhibitor necrostatin-1 (Nec-1): Use of these two inhibitors is considered an effective method to distinguish necroptosis from apoptosis (Kroemer et al. 2009). Thus, Z-VAD and Nec-1 were employed in our study to ascertain the presence and importance of necroptosis (Kaiser et al. 2013).
Materials and Methods
Ethical Approval of the Study Protocol
The study protocol was approved by the animal care committee of Shanghai Jiao Tong University School of Medicine (Shanghai, China). All procedures adhered to the US National Institutes of Health (Bethesda, MD, USA) guidelines for animal experimentation. Appropriate measures were implemented to minimize the suffering of animals, also to minimize the animal number that needed.
Primary Culture of Hippocampal Neurons
As done in our previous studies (Yan et al. 2014), hippocampal neurons were isolated from Sprague Dawley rat embryos at 18 days of pregnancy. Neuron culture medium and experimental consumables were purchased from Life Technologies (Carlsbad, CA, USA). First, hippocampi were dissected from fetal rat brains in cold Hank’s balanced salt solution with no Mg2+ and Ca2+. Then, each hippocampus was dissociated using 0.25% trypsin for enzymatic digestion. Finally, each hippocampus was neutralized with fetal bovine serum at 10% concentration. Isolated hippocampal neurons were then fitted into 6-well plates (Greiner Bio-One, Kremsmünster, Austria) or poly-d-lysine-coated coverslips (Sigma–Aldrich, Saint Louis, MO, USA). Neurons were cultured in serum-free Neurobasal® medium supplemented with 2% B27, 0.5 mM GlutaMAX™ and penicillin -streptomycin (100 U/mL and 100 µg/mL, respectively)) in a 5% CO2 humidified atmosphere at 37 °C. Neurons were plated at a concentration of 1 × 106 cells/cm2. Medium was replaced 6 h after seeding. One-half of the culture medium was replenished on the day 4. Moreover, the culture medium was supplemented with 25 µM l-glutamate (Sigma–Aldrich) during the first 4 days in culture. Cells were pretreated with Nec-1 (10 µM) (catalog number 11658; Cayman Chemical, Ann Arbor, MI, USA) and Z-VAD (ALX-260-138; Alexis Biochemicals, San Diego, CA, USA) in combination or with Z-VAD, Nec-1 alone.
Animals
We used Sprague–Dawley (SD rat) rat pups postnatal day 7 (PND7), weighted 14–18 g purchased from the Animal Center of the Shanghai Jiao Tong University School of Medicine. Rats were fed in cages at 25 °C under a 12-h light–dark cycle (8 am to 8 pm). We administered drugs in a thermostatic container maintained at 36.7 °C, with 65% humidity, as previously published literature (Yan et al. 2014).
A total of 100 rats were used in the present study. After anesthesia, 30 rats were decapitated and each hippocampus was collected for western blotting. A different group of 30 rats was then sacrificed after anesthetized, and brain tissues were dissected for immunohistochemical analyses. The final group of 40 rats was subject to the Morris water maze test (MWM) on day 30 after birth. To prevent potential uncertainty caused by the estrous cycle, only male rats were included in the behavioral testing. In the control group, dimethyl sulfoxide (DMSO) was intraperitoneally injected into the rats. In the sevoflurane group, rats were also intraperitoneally injected with DMSO. In the Z-VAD and Nec-1 groups, rats were, respectively, with intraperitoneal injection of Z-VAD 3 mg/kg or Nec-1 1 mg/kg dissolved in DMSO (Trichonas et al. 2010).
Sevoflurane Exposure
As our previous study (Lu et al. 2016), before sevoflurane exposure, rat pups were separated from mother rats in order to acclimatize to the environment. Pups from the same mother were allocated randomly into one of four groups. The rat pups were assigned randomly to the control group or one of the intervention groups (sevoflurane + DMSO; sevoflurane + Z-VAD; sevoflurane + Nec-1; sevoflurane plus Z-VAD + Nec-1). PND7 rats in the control group received 100% oxygen for 6 h in a thermostatic container at 37 °C. The sevoflurane group (Sev group) rat pups were exposed to 3% sevoflurane (SEVOFRANE®, Osaka, Japan) for 6 h under 100% oxygen in the same thermostatic container at 37 °C. Rat pups in the sevoflurane and control groups received 0.1 mL of DMSO solution (15 μL of DMSO dissolved in 1 mL of physiologic saline), which was the vehicle of Z-VAD and Nec-1. A vaporizer monitored the concentration of sevoflurane in the container. Gas flow into the container was adjusted to 2 L/min. In the intervention studies, mice were administered Z-VAD and/or Nec-1 via intraperitoneal administration 30 min before sevoflurane anesthesia. Doses of Z-VAD and Nec-1 were based on recent publication.14 In Z-VAD and Nec-1 groups, Nec-1 was intraperitoneally injected 10 min after Z-VAD administration.
Western Blotting
Total proteins were extracted from cells using the M-PER mammalian protein extraction reagent (78,503, thermofisher, IL, USA). Equal amounts of total protein (16 μg) were loaded onto 11% SDS-PAGE gels and transferred onto nitrocellulose membranes. The blots were probed with the primary antibodies against RIP1 (1:400), RIP3 (1:600), GAPDH (1:1200) (ab106393, ab62344, ab181602, Abcam, Cambridge, UK), and MLKL (1:800), p-MLKL (1:800) (PA5-105,678, PA5-102,810, Invitrogen, California, UK) followed by probing with the secondary HRP-conjugated goat anti-rabbit antibody (ab6271, Abcam). After washing, the bands were detected by chemiluminescence and imaged with X-ray films. The software “Totallab” was used to scan the optical density of target bands, GAPDH was used as an endogenous reference for normalization.
Co-immunoprecipitation
Cells were lysed in RIPA Lysis and Extraction Buffer (89,900, thermofisher) containing 50 mM phenyl methylsulphonyl fluoride, 0.1 mg/mL chymostatin, 2.5 mM sodium orthovanadate 2 mg/mL aprotinin and 1 mg/mL pepstatin A. After centrifugation at 12,000 g for 5 min, the supernatant was incubated with 5 μL of the rabbit polyclonal to RIP3 primary antibody (sc-374639, Santacruz, CA, USA) for 1 h at 4 °C. The immunocomplex was precipitated from solution using protein A/G magnetic beads and separated by 11% SDS-polyacrylamide gel electrophoresis by following the kit instructions (89,904, thermofisher). Western blotting for RIP1 and PIP3 were performed according to the conventional method.
Hematoxylin and Eosin (H&E) Staining
The rat pups were first perfused with 4% (w/v) paraformaldehyde. After killing, the brains were removed and placed into 20% (w/v) sucrose and finally sectioned into the thickness of 10 µm for the H&E staining. Briefly, we first deparaffinized the sections with xylene and dehydrated. Hematoxylin staining was undertaken for time of 5 min, then washing in tap water with a 10 s rinse in 0.1% HCl solution. Washing in tap water was performed for a second, then the brain sections were stained in eosin solution for 2 min, then washed in tap water again and dehydrated and cleared. At last, for microscopic examination, the brain sections were mounted in neutral gum.
Viability of Neuronal Cells
The viability of neuronal cells was evaluated using the MTT assay. In brief, we changed the culture medium to the Dulbecco’s modified Eagle’s medium (DMEM) containing 0.5 g/L MTT. The supernatant was discarded and the neurons were mixed thoroughly with DMSO (100 μL/well) after incubation for 3 h at 37 °C. When the crystals had dissolved, the absorbance was assessed with an Elx800 plate reader (Bio-Tek Instruments, Winooski, VT, USA) at 570 nm. And Cell viability is measured by absorbance value.
Immunofluorescent Staining
We labeled the hippocampal neurons using the immunofluorescence staining to determine the number of hippocampal neurons. We first cultured the hippocampal neurons for 5 days, and then, fixed the neurons with 4% paraformaldehyde for fifteen minutes followed by incubating overnight with a neuron-specific primary antibody (1:200 dilution; Abcam, Cambridge‚ UK) against neuron-specific enolase and finally the secondary antibody was added to tag the fluorophore. In order to label all the cell nucleus, the cultures were co-stained with 4′,6-diamidino-2-phenylindole (DAPI). Finally, we imaged the neurons by the fluorescence microscope. The content of neurons was assessed by comparing the number of double stained cells with and the number of single stained with only DAPI in each optical field.
MWMT
Thirty days after birth, in order to assess spatial learning and memory ability of the rats, MWMT was conducted. The water maze consisted of a 150 cm diameter circular pool and a video motion tracking system. The pool was divided into four quadrants with two perpendicular lines, and a 13 cm diameter escape platform was placed 2 cm under the surface of the water in one of the quadrants. Warm water added with black ink filled with the pool so as to hide the platform. We tested the rats in the MWM for 6 consecutive days. In order to estimate spatial learning and memory of the rats, positioning navigation test was conducted on day 1 to day 5. The probe trial sessions were carried out 4 times a day from 10:00 to 12:00 for 5 days. Rats were put in the water face to the pool wall and swim freely to search for the platform hidden below water. Escape latency was videotaped by the video motion tracking system. And the escape latency was limited to 90 s. Rats remained on the platform for 10 s. The escape latency was recorded as 90 s if a rat failed to find the platform in 90 s. Then the rats were guided to the platform and standed for 3 s. On day 6, the spatial probe test was carried out to test the memory of the platform space after learning to look for the platform. We removed the platform after completing the positioning navigation experiment. After removing the platform from pool, rats were placed in the quadrant opposite the platform quadrant, then they were allowed to swim freely for 90 s. The time spent in the platform quadrant was recorded. The recording is to assess the ability of spatial position memory.
Statistical Analyses
Data are the mean ± SEM. One-way ANOVA was carried out to determine significant differences among treatment groups using Prism 5 (GraphPad, San Diego, CA, USA). Post hoc comparisons of individual values of mean ± SEM were conducted by Tukey's multiple-comparison tests. P < 0.05 was considered significant.
Results
Sevoflurane-Induced Neurotoxicity and Inhibited the Viabilities in Hippocampal Neurons and Tissue, and the Blockade of Apoptosis or Necroptosis Alone Did Not Attenuate Neurotoxicity or Increase Cell Viability
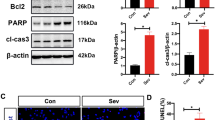
PND7 rats and hippocampal neurons exposed to 3% sevoflurane for 6 h have been shown to undergo neurotoxicity and led to cognitive impairment in a previous study.15 In the present research, it was also reported that exposure to 3% sevoflurane for 6 h induced neurotoxicity and inhibited cell viability (Fig. 1a, b). The next goal was to rescue sevoflurane-induced neurotoxicity (SIN) by introducing the pan-caspase inhibitor Z-VAD to inhibit apoptosis and Nec-1 to inhibit necroptosis.

Sevoflurane-induced neurotoxicity and inhibited the cell viabilities in hippocampal neurons, blockage of apoptosis did not attenuate the neurotoxicity and cell viabilities. a–e Hippocampal neurons lost their normal morphology and detached form the plates in both the SEV group, SEV + Z-VAD group and SEV + Nec-1 group. SEV group exhibited both swelling (black arrow) and shrinkage (red arrow) morphological changes, the SEV + Z-VAD group exhibited only swelling morphological changes (black arrow) and the SEV + Nec-1 group exhibited only shrinkage morphological changes (red arrow). The SEV + Z-VAD + Nec-1 group kept their normal neurons, morphology. f The neuron cell viability assessed by the MTT showed the neuron viability was significant low in SEV group, SEV + Z-VAD group and SEV + Nec-1 group but was rescued to the level of control group in SEV + Z-VAD + Nec-1 group
Interestingly, it was found that inhibiting apoptosis or necroptosis alone did not rescue SIN. However, inhibiting both apoptosis and necroptosis did rescue SIN. The MTT assay showed in vitro that neuron viability was significantly lower in the SEV, SEV + Z-VAD and SEV + Nec-1 groups compared to control group (p < 0.01) (Fig. 1f). However, the results indicated that there was no significant difference in cell viability between the control and SEV + Z-VAD + Nec-1 groups (p > 0.05) (Fig. 1f). Cell viability was significantly higher in the SEV + Z-VAD + Nec-1 group than SEV group. Also, there were clear differences in the morphology of hippocampal neurons between the control, SEV, SEV + Z-VAD, SEV + Nec-1 and SEV + Z-VAD + Nec-1 groups. Hippocampal neurons showed abnormal morphology and were detached from the plates in the SEV, SEV + Z-VAD and SEV + Nec-1 groups. However, there were almost no changes in the morphology of hippocampal neurons in the control and SEV + Z-VAD + Nec-1 groups. The SEV group exhibited swelling (Fig. 1b, black arrow) and shrinkage (red arrow), which indicating necroptosis and apoptosis, whereas the SEV + Z-VAD group exhibited only swelling (Fig. 1c, black arrow), the SEV + Nec-1 group exhibited only shrinkage (Fig. 1d, red arrow).
Hematoxylin and eosin (H&E) of brain tissue in vivo showed the dentate gyrus (Fig. 2, blue-lined box), CA1 area (Fig. 2, black-lined box) and CA3 area (Fig. 2, red-lined box) of the hippocampus of PND7 rats. The SEV group showed swollen dead cells (Fig. 2, black arrow) and shrunken dead cells (Fig. 2, red arrow). The SEV + Z-VAD group showed only swollen dead cells (Fig. 2c, black arrow) in all three areas. The SEV + Nec-1 group showed only shrunken dead cells (Fig. 2d, red arrow) in all three areas. The number of cells underwent necroptosis and apoptosis was counted in three microscopic fields in the dentate gyrus, CA1 area and CA3 area (Fig. 2f and g). The number of cells that underwent necroptosis (p < 0.05) and apoptosis in hippocampal tissues (p < 0.05) was significantly higher after exposure to 3% sevoflurane for 6 h, however, Z-VAD inhibited the number of cells that suffered apoptosis (p < 0.05) but significantly increased the number of cells that underwent necroptosis (p < 0.05). Nec-1 inhibited the number of cells that suffered necroptosis (p < 0.05).

Sevoflurane exposure-induced neurotoxicity in hippocampal tissues, blockage of apoptosis or necroptosis alone did not attenuate the neurotoxicity, inhibiting both apoptosis and necroptosis rescue the sevoflurane-induced neurotoxicity. a–e the HE staining of the brain tissue showed the DG area (blue-lined box), CA1 area (black-lined box) and CA3 area (red-lined box) of the hippocampus of the PND7 rats. SEV group showed both swelling (black arrow) and shrinkage (red arrow) cell death and the SEV + Z-VAD group showed only swelling cell death (black arrow) while the SEV + Z-VAD + Nec-1 group were similar to the control group. f, g The necroptosis and apoptosis cell were calculated by cell morphology in three fields of microscope in DG, CA1 and CA3 area. Sevoflurane exposure increased both necroptosis and apoptosis cell, Z-VAD effectively inhibited the apoptosis cells but significantly increased the necroptosis cells. Nec-1 effectively inhibited the necroptosis cells but significantly increased the apoptosis cells. SEV + Z-VAD + Nec-1 group showed no difference between control group
RIPK1/RIPK3-Mediated Necroptosis was Involved in SIN in Hippocampal Neurons, and Blockade of Apoptosis Enhanced Necroptosis
To further demonstrate the presence of necroptosis and explore the role of necroptosis in SIN, expressions of the necroptosis-related proteins RIPK1, RIPK3 and p-MLKL were quantified in hippocampal neurons via western blotting. Compared to the control group, expression of RIPK1 increased significantly in the SEV and SEV + Z-VAD groups, but not in the control group, SEV + Nec-1 group or SEV + Z-VAD + Nec-1 group (Fig. 3a). Compared with control group, expression of RIPK3 protein was increased significantly in SEV, SEV + Z-VAD and SEV + Z-VAD + Nec-1 groups.

The protein level and binding ability of necroptosis-related protein RIPK1 and RIPK3 in hippocampal neurons. a Western-blot results of the RIPK1, RIPK3 protein and MLKL, p-MLKL protein level. b Co-IP results showed that sevoflurane exposure significantly enhance the binding of RIPK1 and RIPK3 protein. Z-VAD further increased the binding of RIPK1 and RIPK3. The binding capacity returned to the control group level in SEV + Z-VAD + Nec-1 group
Binding of RIPK1/RIPK3 was detected using co-immunoprecipitation (Co-IP). Co-IP results showed that sevoflurane exposure enhanced the binding of RIPK1/RIPK3 protein significantly. However, when apoptosis was inhibited by Z-VAD, the binding of RIPK1/RIPK3 was increased even more significantly (Fig. 3b). These results suggest that necroptosis was involved in SIN in hippocampal neurons, and that binding of RIPK1/RIPK3 is essential for necroptosis. Also, inhibition of apoptosis might enhance the binding, instead of the expression of RIPK1/RIPK3, thus, enhancing necroptosis.
After binding of RIPK1/RIPK3, necroptosis is activated by the phosphorylation of mixed lineage kinase domain-like (p-MLKL).So we quantified expression of p-MLKL by western blotting. The expression of p-MLKL significantly increased in the SEV and SEV + Z-VAD groups, but not in the SEV + Nec-1 group or SEV + Z-VAD + Nec-1 group compared to the control group (Fig. 3a).
Sevoflurane Increased the Expression of RIPK1 and RIPK3 in Hippocampal Tissues In Vivo
To further explore RIPK1/RIPK3-mediated necroptosis in vivo, immunofluorescence was used to reveal expression of RIPK1 and RIPK3 protein and their location in the hippocampal tissues of PND7 rats. After exposure to 3% sevoflurane for 6 h, expression of the RIPK1 (Fig. 4I/b,e) and RIPK3 (Fig. 4II/b,e) was noted in the hippocampal tissues of PND7 rats, whereas barely any expression of RIPK1 (Fig. 4I/I) and RIPK3 (Fig. 4II/I)was detected in the control group.

Immunofluorescence showed the expression of RIPK1 and RIPK3 protein and its location in the hippocampal tissues of the PND7 rats. a RIPK1 protein. b RIPK3 protein. (I) Immunofluorescence of RIPK1 protein in the hippocampal tissues. (II) Immunofluorescence of RIPK3 protein in the hippocampal tissues. The color boxes (red, blue, green) in the second row (D, E, F) is the magnification of the first row (A, B, C), respectively
Inhibition of Apoptosis and Necroptosis Reduced Sevoflurane-Induced Cognitive Deficiency Significantly
The MWM test was conducted for evaluation of learning and memory in the rats. Compared to the control group, the distance center-point total was significantly higher in the SEV group on days 3–5 and in the SEV + Z-VAD, SEV + Nec-1 groups on day 3, (p < 0.05) (Fig. 5a), but no differences were found between the SEV + Z-VAD + Nec-1 group and control group. In zone 5, the escape latency was significantly higher in the SEV group on days 3–5 and in the SEV + Z-VAD, SEV + Nec-1 groups on day 3, compared to the control group (p < 0.05) (Fig. 5b). Time spent in the target quadrant was significantly shorter in the SEV group (p = 0.001) and SEV + Z-VAD group (p = 0.028), SEV + Nec-1 group (p = 0.025) compared to the control group (Fig. 5c). No differences was found between the SEV + Z-VAD + Nec-1 and control groups. Heatmaps and tracked swimming changes indicated that rats in the SEV, SEV + Z-VADand SEV + Nec-1 groups spent less time in the target quadrant compared with rats in the control group (Fig. 5d) and there was no significant difference between the SEV + Z-VAD + Nec-1 and control groups (Fig. 5d). Taken together, these data showed that the blockade of apoptosis and necroptosis attenuated sevoflurane-induced impairment of cognitive function in young rats.

Block both of apoptosis and necroptosis alleviated the sevoflurane-induced cognitive function impairment in the rats. The Morris water maze test was used to assess spatial learning and memory ability of 30 day old rats (n = 8). a Distance center-point total (*p < 0.05 vs. Control group). b In zone5 latency to first *p < 0.05 vs. Control group). c Time in target quadrant (**p = 0.001 vs. Control group, *p = 0.028 vs. Control group). d Heatmap of time in the target quadrant
Discussion
There have been many investigations of anesthesia-induced neurotoxicity in the developing brain of neonates in animals and humans. Sevoflurane is the most commonly used inhalation anesthetic.
Here, it was shown that sevoflurane exposure elicited neurotoxicity in hippocampal neurons and tissues. Also, it was also shown that the blockade of apoptosis or necroptosis alone did not attenuate SIN and cell viability. Furthermore, it was revealed that RIPK1/RIPK3-mediated necroptosis was involved in SIN in hippocampal neurons, and that the blockade of apoptosis enhanced necroptosis. Finally, it was found that inhibition of both apoptosis and necroptosis rescued SIN. These data offer potential new ideas indications for clinical anesthesia regarding the prevention of neurotoxicity caused by anesthesia exposure in neonates.
Infants are administered general anesthesia for diagnostic and surgical procedures. The safety of anesthesia in neonates and infants is controversial. Accumulating evidence from animal and clinical studies (Wilder et al. 2009; DiMaggio et al. 2011; Ing et al. 2012; Hansen et al. 2011; Sun 2010.) has suggested that exposure to anesthetics in the neurodevelopmental period can result in neurotoxicity and learning difficulties (Deng et al. 2014; Zou et al. 2011). Sevoflurane exposure has been shown to lead to neuronal apoptosis and pathologic alterations in the hippocampus of neonatal rats (Feng et al. 2012; Zhang et al. 2008; Satomoto et al. 2009). However, the mechanisms underlying SIN are largely unknown.
Importantly, it was found that, except for apoptosis, necroptosis (a novel manner pathway of cell death) was involved in the mechanisms underlying SIN. It was also found that necroptosis might act as an “insurance policy” if apoptosis is inhibited. To prevent SIN, inhibiting apoptosis or necroptosis alone was demonstrated to be insufficient in our study. Blockade of both apoptosis and necroptosis may provide new thinking for preventing SIN in neonates.
Necroptosis is a newly confirmed type of programmed cell death. It is essential for normal embryonic development, chronic intestinal inflammation, and T-cell proliferation (Peter 2011; Kaiser et al. 2011.). Necroptosis is different from apoptosis, since it is a caspase-independent process. Also, it is different from conventional necrosis because necrosis is a type of unexpected cell death whereas necroptosis is a tightly controlled process of cell death. Necroptosis is largely dependent on RIPK1/RIPK3 and MLKL, and formation of RIPK1/RIPK3 complexes is essential for necroptosis. Necroptosis can be blocked by the RIPK1 inhibitor Nec-1. It was shown that the inhibition of apoptosis significantly increased RIPK1/RIPK3 expression and enhanced the binding of RIPK1/RIPK3, which indirectly demonstrates the existence of necroptosis.
Studies have shown that necroptosis may act as an alternate pathway if apoptosis or necrosis are inhibited. For example, Moon and colleagues have investigated on the regulation of necrosis and apoptosis after spinal-cord injury (Moon et al. 2012). They found that the protection against the death of neurocytes via inhibition of necrosis and apoptosis is limited. Additionally, Liu and colleagues indicated that necroptosis contributed to the death of nerve cells and affected functional outcomes in mice that with suffered spinal-cord injuries (Liu et al. 2015). Inhibiting necroptosis via Nec-1 administration may serve as a feasible therapeutic strategy to treat spinal-cord injury. In this study, the same situation was applied in SIN.
It was found that apoptosis and necroptosis coexisted in SIN and that necroptosis may act as an alternate cell death pathway if apoptosis is inhibited. It was hypothesized that the inhibition of apoptosis and necroptosis may rescue SIN. Hence, the pan-caspase inhibitor Z-VAD and RIPK1 inhibitor Nec-1 were introduced to test this hypothesis.
The MTT assay in vitro assay showed that neuron viability was rescued to a more closely observed level in the control group compared to the SEV + Z-VAD + Nec-1 group (Fig. 1f). H&E staining of brain tissue in vivo showed that the SEV + Z-VAD + Nec-1 group was similar to control group (Fig. 2e). The number of cells that underwent necroptosis and apoptosis was counted in three microscopic fields: the SEV + Z-VAD + Nec-1 group showed no significant difference compared to the control group (Fig. 2f, g). In terms of expression and binding of RIPK1/RIPK3, the SEV + Z-VAD + Nec-1 group showed obviously low RIPK3 expression compared with that in the SEV + Z-VAD group, but no significant difference in expression of RIPK1 protein was noted. Co-IP showed that the binding capacity of the RIPK1/RIPK3 protein returned to that observed in the control group in the SEV + Z-VAD + Nec-1 group (Fig. 4). Taken together, the results shown above suggest the inhibition of apoptosis or necroptosis alone provided only a limited effect against SIN, whereas inhibition of both apoptosis and necroptosis played an important protective role.
The study had four main limitations. First, due to sevoflurane diverse effects, it is difficult to distinguish key factor leading to its neurotoxicity properties. Second, the other effects of sevoflurane were not detected, such as anesthesia-related cerebral anoxia, which may have affected the results. Therefore, further experiments are needed to explore the roles of these effects. Third, other brain regions related to learning and memory (e.g., the amygdala and striatum) were not included in this study, but should can be considered in future research. Finally, animal experiments cannot be directly converted into clinical conclusions applied to humans.
Conclusions
Sevoflurane exposure elicited neurotoxicity in neonatal hippocampal neurons and tissues. The blockade of apoptosis alone did not attenuate SIN. RIPK1/RIPK3-mediated necroptosis was involved in SIN in hippocampal neurons. SIN could be attenuated only by inhibiting apoptosis and necroptosis.
Abbreviations
- Z-VAD:
-
ZVAD-fmk
- Nec-1:
-
Necrostatin-1
- RIPK1:
-
Receptor-interacting protein kinase 1
- RIPK3:
-
Receptor-interacting protein kinase 3
- FDA:
-
Food and Drug Administration
- GABA:
-
Gamma-aminobutyric acid
- NMDA:
-
N-methyl-d-aspartate
- MLKL:
-
Mixed lineage kinase domain-like
- FBS:
-
Fetal bovine serum
- DMSO:
-
Dimethyl sulfoxide
- TBST:
-
Tris buffer solution Tween
- HE:
-
Hematoxylin staining
- MTT:
-
Methyl thiazolyl tetrazolium
- NSE:
-
Neuron-specific enolase
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- SEV:
-
Sevoflurane
- Co-IP:
-
Co-immunoprecipitation
References
Andropoulos DB, Greene MF (2017) Anesthesia and developing brains - implications of the FDA warning. N Engl J Med 376:905
Berghe TV, Linkermann A, Jouan-Lanhouet S et al (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 15:134–146
Cai Z, Jitkaew S, Zhao J et al (2014) Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16:55–65
Davidson A, Vutskits L (2017) The new FDA drug safety communication on the use of general anesthetics in young children: what should we make of it? Paediatr Anaesth 27:336–337
De Almagro MC, Vucic D (2015) Necroptosis: pathway diversity and characteristics. Semin Cell Dev Biol 39:56–62
Deng M, Hofacer RD, Jiang C et al (2014) Brain regional vulnerability to anesthesia-induced neuroapoptosis shifts with age at exposure and extends into adulthood for some regions. Br J Anaesth 113:443–451
DiMaggio C, Sun LS, Li G (2011) Early childhood exposure to anesthesia and risk of developmental and behavioral disorders in a sibling birth cohort. Anesth Analg 113:1143–1151
Feng X, Liu JJ, Zhou X et al (2012) Single sevoflurane exposure decreases neuronal nitric oxide synthase levels in the hippocampus of developing rats. Br J Anaesth 109:225–233
Hansen TG, Pedersen JK, Henneberg SW et al (2011) Academic performance in adolescence after inguinal hernia repair in infancy: a nationwide cohort study. Anesthesiology 114:1076–1085
Huang Z, Wu SQ, Liang Y et al (2015) RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe 17:229–242
Ing C, DiMaggio C, Whitehouse A et al (2012) Long-term differences in language and cognitive function after childhood exposure to anesthesia. Pediatrics 130:e476–e485
Kaiser WJ, Upton JW, Long AB et al (2011) RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471:368–372
Kaiser WJ, Sridharan H, Huang C et al (2013) Toll like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288:31268–31279
Khoury MK, Gupta K, Franco SR et al (2020) Necroptosis in the pathophysiology of disease. Am J Pathol 190:272–285
Kroemer G, Galluzzi L, Vandenabeele P et al (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16:3–11
Linkermann A, Green DR (2014) Necroptosis. N Engl J Med 370:455–465
Liu Q, Qiu J, Liang M et al (2014) Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis 5:e1084
Liu M, Wu W, Li H et al (2015) Necroptosis, a novel type of programmed cell death, contributes to early neural cells damage after spinal cord injury in adult mice. J Spinal Cord Med 38:745–753
Lu Y, Wu X, Dong Y et al (2010) Anesthetic sevoflurane causes neurotoxicity differently in neonatal naive and Alzheimer disease transgenic mice. Anesthesiology 112(6):1404–1416
Lu Y, Huang Y, Jiang J et al (2016) Neuronal apoptosis may not contribute to the long-term cognitive dysfunction induced by a brief exposure to 2% sevoflurane in developing rats. Biomed Pharmacother 78:322–328
Moon YJ, Lee JY, Oh MS et al (2012) Inhibition of inflammation and oxidative stress by angelica dahuricae radix extract decreases apoptotic cell death and improves functional recovery after spinal cord injury. J Neurosci Res 90:243–256
Moriwaki K, Bertin J, Gough PJ et al (2015) Differential roles of RIPK1 and RIPK3 in TNF induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis 12:e1636
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517:311–320
Peter ME (2011) Programmed cell death: apoptosis meets necrosis. Nature 471:310–312
Satomoto M, Satoh Y, Terui K et al (2009) Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology 110:628–637
Sun L (2010) Early childhood general anaesthesia exposure and neurocognitive development. Br J Anaesth 105(Suppl 1):i61–i68
Trichonas G, Murakami Y, Thanos A et al (2010) Receptor interacting protein kinases mediate retinal detachment-induced photoreceptor necrosis and compensate for inhibition of apoptosis. Proc Natl Acad Sci USA 107:21695–21700
Vandenabeele P, Galluzzi L, Berghe TV et al (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714
Wilder RT, Flick RP, Sprung J et al (2009) Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology 110:796–804
Xiao H, Liu B, Chen Y et al (2016) Learning, memory and synaptic plasticity in hippocampus in rats exposed to sevoflurane. Int J Dev Neurosci 48:38–49
Yan J, Huang Y, Lu Y et al (2014) Repeated administration of ketamine can induce hippocampal neurodegeneration and long-term cognitive impairment via the ROS/HIF-1α pathway in developing rats. Cell Physiol Biochem 33:1715–1732
Zhang X, Xue Z, Sun A (2008) Subclinical concentration of sevoflurane potentiates neuronal apoptosis in the developing C57BL/6 mouse brain. Neurosci Lett 447:109–114
Zou X, Liu F, Zhang X et al (2011) Inhalation anesthetic-induced neuronal damage in the developing rhesus monkey. Neurotoxicol Teratol 33:592–597
Funding
This research was funded by Grant of National Natural Science Foundation 81901093 (to Y.L.) and National Natural Science Foundation 81671059 (to Y.L.).
Author information
Authors and Affiliations
Contributions
Conceptualization, YL and WL; methodology, RX and YZ; validation, JJ; formal analysis, WL; data curation, RX; writing—original draft preparation, RX and YZ; writing—review and editing, YL and WL; funding YL.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Xu, R., Zhu, Y., Jia, J. et al. RIPK1/RIPK3-Mediated Necroptosis is Involved in Sevoflurane-Induced Neonatal Neurotoxicity in the Rat Hippocampus. Cell Mol Neurobiol 42, 2235–2244 (2022). https://doi.org/10.1007/s10571-021-01098-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-021-01098-z