
Abstract
An increasing number of studies have found that use of traditional anesthetics may lead to cognitive impairment of the immature brain. Our previous studies verified that cyclin-dependent kinase 5 (CDK5) plays a role in sevoflurane-induced cognitive dysfunction. Autophagy was shown to protect against anesthesia-induced nerve injury. Therefore, the current study aimed to ascertain if autophagy participates in anesthesia-induced neurotoxicity. In this study, primary hippocampal neurons were isolated and utilized for experiments in vitro. We also performed in vivo experiments with 6-day-old wild-type mice treated with or without roscovitine (Rosc, a CDK5 inhibitor) or 3-methyladenine (3-Ma, an autophagy inhibitor) after exposure to sevoflurane. We used the Morris water maze to analyze cognitive function. Immunohistochemical staining was used to assess pathologic changes in the hippocampus. The results showed that suppressing CDK5 reversed sevoflurane-induced nerve cell apoptosis both in vivo and in vitro and demonstrated that inhibits CDK5 activation promoted Sirtuin 1 (Sirt1) expression, which functions importantly in induced autophagy activation. Suppression of Sirt1 expression inhibited the protective effect of Rosc on sevoflurane-induced nerve injury by inhibiting autophagy activation. Our in vivo experiments also found that pretreatment with 3-Ma attenuated the protective effect of Rosc on sevoflurane-induced nerve injury and cognitive dysfunction. We conclude that inhibits CDK5 activation restored sevoflurane-induced cognitive dysfunction by promoting Sirt1-mediated autophagy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sevoflurane is the most common inhaled anesthetic in surgery (Ghatge et al. 2003). However, an increasing number of studies have found that sevoflurane-induced cytotoxicity may lead to cognitive impairment at an early age in both humans and animals (Alkire et al. 2008; Wiklund et al. 2009; Rohan et al. 2005). Previous investigations have observed that the sevoflurane-induced stress environment can lead to neural neuroinflammation, neuronal apoptosis and abnormal protein deposition which contributes to the cognitive impairment (Ge et al. 2015; Lin and Zuo 2011; Shen et al. 2013).
Autophagy is an evolutionarily conserved process of self-digestion, which functions to maintain cellular homeostasis (Glick et al. 2010). It was found that excessive autophagy contributes to neuronal apoptosis in a stress environment (Xiao et al. 2017). At gestational day 14, sevoflurane exposure induced activation of PTEN/Akt/mTOR pathway-mediated autophagy in the fetal brain resulting in sevoflurane-induced neurotoxicity. Autophagy inhibition reversed anesthesia-induced neural stem cell apoptosis, proliferation decline and memory deficits (Li et al. 2017). However, other investigations have reported that a certain amount of autophagy activation ameliorated cognitive impairment (Guo et al. 2018; Zhang et al. 2018b; Zhou et al. 2016). Therefore, whether autophagy has a protective effect in the developing brain and ameliorates sevoflurane-induced toxicity is still unclear.
Cyclin-dependent kinase 5 (CDK5) functions in neural development and forms complexes with p39 or p35 (Zheng et al. 2016). Our previous study showed that inhibiting CDK5 with roscovitine (Rosc) can effectively inhibit sevoflurane anesthesia-induced neuronal injury and cognitive dysfunction via regulation of ERK/PPARγ/CREB and Tau/GSK3β signaling (Liu et al. 2017b). It is also known that CDK5 decreases Sirtuin 1 (Sirt1) expression (Zhang et al. 2018a). Sirt1 has deacetylase activity which is dependent on nicotinamide adenine dinucleotide (NAD+) and belongs to the class III histone deacetylase family (Stunkel and Campbell 2011; Michan and Sinclair 2007). Increasing evidence has demonstrated that Sirt1 has indispensable functions in stress responses, senescence, and regulation of cellular metabolism (Ou et al. 2014; Chang and Guarente 2013). Studies have also indicated that expression of Sirt1 is involved in autophagy activation (Sun et al. 2018; Fan et al. 2017).
The current study aimed to investigate if autophagy plays a role in sevoflurane-induced cognitive dysfunction in fetal mice. We also studied the regulatory relationships among CDK5, Sirt1, autophagy, and sevoflurane-induced cytotoxicity.
Materials and Methods
Animals
Pregnant mice were obtained (Fudan University Animal Care Committee, Shanghai, China) at least 1 week prior to experiments in order to adapt to the laboratory environment and were housed in a standard laboratory environment with free access to food and tap water under a 12:12-h light/dark cycle at 22 °C with 60% humidity. Shanghai Tongji University School of Medicine Animal Care and Use Committee (Shanghai, China) approved all animal experiments.
Anesthesia Exposure
We used 7-day-old male mice for this study and randomly divided them into control and sevoflurane treatment groups. We exposed mice in the sevoflurane treatment group to 2.2% sevoflurane (~ 1.0 minimum alveolar concentration) for 3 consecutive days for 2 h/day. We continuously monitored anesthetic, O2 and CO2 concentrations in the chamber (GE Datex 5 Ohmeda; Soma Technology, Tewksbury, MA, USA). Body temperature was maintained between 37 °C and 38 °C with a warming blanket. After three consecutive days of anesthetic, we euthanatized some mice in each group and extracted and froze their brains in liquid nitrogen for further study. The remaining mice in each group were used for memory and learning function testing after growth to an appropriate age.
To evaluate if CDK5 played a role in sevoflurane-induced hippocampal neuronal injury, CDK5 inhibitor Rosc (25 mg/kg) was injected intraperitoneally before sevoflurane exposure. In order to identify if autophagy was involved in sevoflurane-induced hippocampal neuronal injury, autophagy inhibitor 3-methyladenine (3-Ma; 1.5 mL/kg in a 10% phosphate-buffered saline (PBS) solution) was injected intraperitoneally before sevoflurane anesthesia.
Morris Water Maze Test
Memory and learning functions were tested in a Morris water maze (MWM) (Lv et al. 2017). One operator blinded to the treatment groups carried out the testing. The apparatus consisted of a round steel pool (height 60 cm and diameter 122 cm) that was filled with water to a level 1 cm higher than a platform (depth 30 cm and diameter 10 cm). The pool was surrounded by a blue curtain with cues and was located in an isolated room (60% humidity, 20 °C). We opacified the water by adding titanium dioxide and maintained the water at 21 °C.
Testing continued for 5 days after initiation on P40. The first 4 days (P40–P43) were used for reference memory tests. All mice were trained in four trials every day for 6 days with an inter-trial interval of 30–40 min. Prior to each trial, we placed the mouse at different starting positions in the water facing the wall. We allowed it to stay on the platform for 15 s if it was not able to find the platform within 1 min. The swimming activity of each animal was record with a video tracking system. We recorded escape latency, i.e., time from placement in the water to finding the platform. On P44, we removed the platform from the pool and performed a spatial probe test. The mouse was allowed to swim freely for 2 min before being placed in the opposite quadrant. We recorded platform crossing numbers. We analyzed the data using motion detection software designed for the MWM test (Shanghai Mobile Datum Information Technology Co., Shanghai, China).
Immunofluorescence Staining
Brain sections (5 μm) were prepared for immunofluorescence staining. In order to reduce background staining, we used PBS containing 10% fetal bovine serum (FBS) to incubate the brain sections for 30 min at room temperature. The sections were stained by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) to identify apoptotic neurons. We performed LC3 staining to evaluate autophagy. We used a fluorescence microscope (Nikon, Tokyo, Japan) or Axiophot light microscope (Zeiss, Oberkochen, Germany) for photomicrography and histochemical analysis.
Cell Culture and Treatment
We isolated primary hippocampal neurons and cultured them using the method of Liu et al. 2017a. In brief, we isolated the hippocampi from P0 mice and cut them into small pieces before digestion with 0.125% trypsin at 37 °C for 15 min. Then, we seeded cells onto poly-d-lysine (10 mmol/L)-coated 10-mm dishes before trituration and centrifugation and cultured them with neurobasal medium (Gibco, Carlsbad, CA, USA) supplemented with 0.25% Glumax (Gibco) and 2% B27 (Gibco) with 1 × 106 cells/mL. After 3 days, in order to inhibit glial cell proliferation, we added 2.5 μg/mL cytosine arabinoside (Sigma-Aldrich, St. Louis, MO, USA) to the culture medium for 1 day. We replaced 50% of the medium every third day and continuously cultured the cells for 14 days (37 °C with 5% CO2) before the following experiments.
To mimic the in vivo anesthesia conditions induced by sevoflurane in vitro, we replaced the neurobasal medium used for hippocampal neuron cultures with glucose-free neurobasal medium supplemented with 2.78 mmol/L glucose and 1% B27 according to Wang et al. 2014. We exposed hippocampal neurons to 4% sevoflurane for 1 day with/without Rosc (5 μM) treatment (37 °C with 5% CO2) and then cultured them at 37 °C with 5% CO2 for 1 additional day. We identified isolated neurons under immunofluorescence by βIII tubulin staining.
RNA Interference and Overexpression
To detect CDK5 and Sirt1 effects on cellular survival, siRNA against Sirt1 and a CDK5 overexpression vector were synthesized by GenePharma (Shanghai, China). We transfected primary hippocampal neurons using Lipofectamine 2000 (Thermo Scientific, Waltham, MA, USA) following standard procedures.
Flow Cytometry
We detected primary hippocampal neuronal apoptosis by flow cytometry. Briefly, we washed primary hippocampal neurons from each group twice and adjusted them to a concentration of 1 × 106 cells/mL with cold D-Hanks buffer. We then added propidium iodide (PI, 10 μL) and annexin V (AV)-FITC (AV-FITC, 10 μL) to a cell suspension of 100 μL and incubated them at room temperature in the dark for 15 min. Finally, we added 400 μL of binding buffer to each sample and analyzed them using flow cytometry.
Western Blot Analysis
We resolved proteins from hippocampal tissues or cells by SDS–polyacrylamide gel electrophoresis through a 5% stacking gel and a 12% separating gel followed by staining with Coomassie blue to determine the enzyme preparation purity. We performed western blotting as follows. We transferred proteins from the gels onto nitrocellulose membranes by electroblotting and blocked the membranes in TBS-T buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 0.2% Tween-20) containing 5% nonfat milk for 1 h at room temperature. We probed the membranes with antibodies against the proteins listed below (all provided by Santa Cruz Biotechnology, Dallas, TX, USA) to determine protein expression: CDK5 (1:1000), Sirt1 (1:1000), Beclin-1 (1:500), LC3 (1:500), caspase-3 (1:1000), cleaved caspase-3 (1:1000), p-CDK5 (1:1000), and β-actin (1:1000). The blots were then incubated with horseradish peroxidase-conjugated secondary antibody (1:1000, Santa Cruz). We used the ECL chemiluminescent kit (Millipore, Danvers, MA, USA) to visualize the protein bands. Protein abundance was measured by ImageJ 1.48u4 software, which was normalized against the corresponding β-actin loading control.
Cell Viability Analysis
We measured cell viability via the CCK8 assay (cell counting kit-8, 7 sea Molecular Technologies, Shanghai, China) following a standard process. We seeded cells in 96-well cell culture plates at a cellular density of 1 × 104 cells per well. After exposure to different doses of sevoflurane (0%, 1%, 2% and 4%) for 0 h, 24 h, 48 h, and 72 h, the cell monolayers were rinsed with PBS three times. We added CCK8 solution diluted 1:10 in neurobasal medium to the cells for two hours at 37 °C. We measured absorbance by a microplate reader at 450 nm.
Statistical Analysis
Continuous variables are presented as means ± SD (standard deviation). We evaluated statistical significance by analysis of variance followed by Tukey–Kramer multiple comparison tests and Student’s t test. P values ≤ 0.05 were regarded as statistically significant.
Results
Sevoflurane Treatment Promoted Apoptosis of Primary Cultured Hippocampal Neurons
To determine if sevoflurane could induce hippocampal neuron injury, we isolated primary cultured hippocampal neurons from newborn C57BL/6 mouse (postnatal day P0). Primary hippocampal neurons had long dendrites (Fig. 1a). Immunofluorescence assays showed that the isolated cells expressed neuron-specific protein βIII tubulin but not glial cell marker protein GFAP, suggesting that the isolated cells were hippocampal neuronal cells (Fig. 1b). Increasing doses of sevoflurane (1%, 2%, and 4%) were then applied to the hippocampal neurons for different times (24, 48 and 72 h) and CCK8 assays were used to assess cell viability. The results showed that neuronal cell viability decreased as sevoflurane concentration increased (Fig. 1c). Flow cytometry demonstrated that apoptosis of hippocampal neurons was increased after treatment with sevoflurane and the increase in apoptotic rate was concentration-dependent (Fig. 1d, e). Western blots revealed that the cleaved caspase-3 level was increased after exposure to sevoflurane in a dose-dependent manner (Fig. 1f, g).

Sevoflurane treatment promoted apoptosis in primary cultured hippocampal neurons. a Photomicrograph displaying isolated neurons. Scale bar, 50 μm. b Immunofluorescence assay showing expression of neuron-specific protein βIII tubulin and glial-specific protein GFAP in isolated cells. Scale bar, 50 μm. c CCK8 assay shows cell viability after treatment with increasing doses of sevoflurane over time. Data are presented as means ± SD. **P < 0.01, ***P < 0.001 versus control. d Cell apoptosis was analyzed by flow cytometry after double-labeling with annexin V-FITC and PI after treatment with sevoflurane for 24 h. e The percentage of apoptotic cells in each group was analyzed. Data are presented as means ± SD. ***P < 0.001 versus control. f and g The expression of cleaved caspase-3 and total caspase-3 was determined by western blotting. β-actin was used as an endogenous control. The histogram represents means ± SD. ***P < 0.001 versus control
Suppression of CDK5 Activation Decreased the Toxicity of Sevoflurane by Promoting Sirt1-Mediated Autophagy
To examine the possible role of autophagy in neuroprotection, primary hippocampal neurons were pretreated with Rosc. Results illustrated that pretreatment with Rosc significantly suppressed phosphorylation of CDK5 in primary hippocampal neurons and promoted Sirt1 expression. Silencing Sirt1 had no influence on phosphorylation of CDK5 under 4% sevoflurane exposure (Fig. 2a–c). Flow cytometry showed that apoptosis of hippocampal neurons decreased after pretreatment with Rosc but downregulating the expression of Sirt1 reversed the Rosc protective effect against sevoflurane-induced apoptosis (Fig. 2d, e). Western blots also showed that pretreatment with CDK5 inhibitor Rosc decreased the expression of cleaved caspase-3, which then rose when Sirt1 was also silenced (Fig. 2f, g). Immunofluorescence detection found that pretreatment with Rosc significantly activated autophagy, while downregulation of Sirt1 reversed Rosc-induced autophagy after exposure to sevoflurane (Fig. 2h, i), suggesting that Sirt1 functions indispensably in autophagy induction. Western blots further confirmed that downregulation of Sirt1 expression reversed the Rosc-induced promotion of Beclin-1 and LC3 expression during sevoflurane exposure (Fig. 2j–l).

Suppression of CDK5 activation decreased toxicity from sevoflurane by promoting Sirt1-mediated autophagy. Primary neurons were pretreated with 10 μM Rosc for 2 h before 4% sevoflurane treatment for 24 h. a–c Western blots show the expression of CDK5 and Sirt1 in different treatment groups. β-actin was used as an endogenous control. Bars represent means ± SD. ***P < 0.001 versus control. ###P < 0.001 versus 4% sevoflurane + Rosc group. d Cell apoptosis was analyzed by flow cytometry after double-labeling with annexin V-FITC and PI. e The percentage of apoptotic cells in each group was analyzed. Data are presented as means ± SD. ***P < 0.001 versus control. ###P < 0.001 versus 4% sevoflurane-treated group. f and g The expression of cleaved caspase-3 and total caspase-3 was determined by western blotting. β-actin was used as an endogenous control. Bars represent means ± SD (n = 3). ***P < 0.001 versus control. ###P < 0.001 versus 4% sevoflurane-treated group. h and i Immunofluorescence shows autophagic puncta in primary neurons. Bars represent means ± SD. ***P < 0.001 versus control. ###P < 0.001 versus 4% sevoflurane-treated group. Scale bar, 20 μm. j–l Western blots show the expression of LC3 and Beclin-1. Bars represent means ± SD. ***P < 0.001 versus control. ###P < 0.001 versus 4% sevoflurane-treated group

Overexpression of CDK5 promoted the degradation of Sirt1. a and b Western blots show the expression of CDK5 protein after transfection with a CDK5 overexpression vector (CDK5) or negative control (NC) for 48 h. Bars represent means ± SD. ***P < 0.001 versus control. c and d Western blots show the expression of Sirt1 after upregulation of CDK5 in primary neurons. Bars represent means ± SD. ***P < 0.001 versus control
To better detect the CDK5 regulatory effect on Sirt1 expression, a CDK5 overexpression vector was transfected into primary hippocampal neurons for 48 h. Expression of CDK5 was significantly increased compared to non-transfected and negative control-transfected cells (Fig. 3a, b), while upregulation of CDK5 decreased the expression of Sirt1 (Fig. 3c, d).
CDK5 Inhibition Decreased Sevoflurane-Induced Neuronal Apoptosis by Promoting Sirt1-Mediated Autophagy
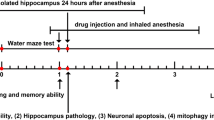
We designed a flow chart of the experiment to completely analyze sevoflurane effects on the neonatal mouse, which is shown in Fig. 4. A range of techniques were employed to analyze both the cellular and molecular changes in neuronal tissue as well as behavioral and phenotypic changes in mice following sevoflurane exposure. P6 mice were exposed to 2.2% sevoflurane three times per day for a total exposure duration of 2 h per day with or without pretreatment with Rosc or 3-Ma. We then performed molecular analyses including western blot analysis and immunofluorescence on hippocampal tissues sampled from the mice on P10 to investigate the interaction between Sirt1-mediated autophagy and neuroprotection in vivo. Results showed that sevoflurane exposure significantly promoted phosphorylation of CDK5, while CDK5 inhibitor pretreatment suppressed the activation of CDK5 (Fig. 5a–c). The results also demonstrated that Rosc pretreatment reversed the sevoflurane-induced inhibition of Sirt1 expression and that autophagy inhibitor 3-Ma pretreatment had no effect on either CDK5 activation or Sirt1 expression (Fig. 5a–c), suggesting that CDK5 and Sirt1 can regulate autophagy. Immunofluorescence staining for TUNEL showed that Rosc pretreatment reversed sevoflurane-induced neuronal apoptosis in the hippocampus, but the protective effect of Rosc was suppressed after pretreatment with 3-Ma (Fig. 5d). Western blots confirmed that suppression of autophagy by 3-Ma reversed the inhibition of cleaved caspase-3 induced by Rosc after sevoflurane exposure (Fig. 5e, f).

Schematic timeline of the experimental design. Postnatal day (P) when procedures/experiments were performed is indicated. IHC immunohistochemistry, IF immunofluorescence, MWM Morris water maze test

Inhibition of CDK5 decreased sevoflurane-induced neuronal apoptosis by promoting Sirt1-mediated autophagy. All experiments were performed using hippocampal tissues of mice exposed to 2.2% sevoflurane and treated with/without Rosc (25 mg/kg) or 10% 3-Ma (1.5 mL/kg) at postnatal day 10. a–c The expression of CDK5 and Sirt1 in hippocampal tissues was detected by western blot. The data are presented as means ± SD. ***P < 0.001 versus control. #P < 0.05, ###P < 0.001 versus Rosc treatment group. d Representative images of immunofluorescent staining for TUNEL. Scale bar, 50 μm. e and f The expression of cleaved caspase-3 was measured by western blot analysis. β-actin was used as an endogenous control. The data are presented as means ± SD. ***P < 0.001 versus control. ###P < 0.001 versus Rosc treatment group. g Immunofluorescence detection shows the autophagic puncta in hippocampal tissues with LC3 stain. Scale bar, 100 μm. h–j Western blots show the expression of LC3 and Beclin-1. Bars represent means ± SD. ***P < 0.001 versus control. ###P < 0.001 versus Rosc treatment group
To further confirm that the Rosc neuroprotective effect was related to autophagy, immunofluorescent staining of LC3 showed that pretreatment with Rosc significantly promoted autophagy activation, but 3-Ma treatment reversed this effect (Fig. 5g). Western blots also confirmed that pretreatment with Rosc promoted Beclin-1 and LC3 expression, while 3-Ma treatment blocked the Rosc effect on these proteins (Fig. 5h, j).
Inhibition of CDK5 Decreased Sevoflurane-Induced Cognitive Impairment by Promoting Autophagy Activation
To examine the cognitive ability of mice following sevoflurane exposure, we performed MWM tests, which are a broadly utilized technology for assessing spatial learning and memory. The result showed that sevoflurane treatment increased escape latencies when compared with the control group (Fig. 6a). Furthermore, suppression of CDK5 activity by pretreatment with Rosc significantly decreased the time percentage spent by the mice in the target quadrant, while 3-Ma treatment suppressed the protective effect of Rosc (Fig. 6a). Spatial probe tests showed that sevoflurane treatment decreased the number of platform crossings comparing with the control group, but inhibiting CDK5 activity increased the number of platform crossings comparing with the control group (Fig. 6b). These data suggested that inhibition of CDK5 activity decreased sevoflurane-induced cognitive impairments by promoting autophagy activation.

Inhibition of CDK5 decreased sevoflurane-induced cognitive impairment. a The mice in the sevoflurane treatment group exhibited significantly longer escape latencies than those in the control group. Downregulation of CDK5 expression significantly decreased the escape latency. The data are presented as means ± SD. ***P < 0.001 versus control. ###P < 0.001 versus sevoflurane group. b Platform crossings were decreased in the sevoflurane treatment group but increased after downregulation of CDK5 expression compared with the control group. The data (n = 5) are presented as means ± SD. *P < 0.05, ***P < 0.001 versus control. ###P < 0.001 versus sevoflurane
Discussion
The present investigation evaluated the role of Sirt1 in sevoflurane-induced nerve injury. Results confirmed that sevoflurane exposure promoted CDK5 activation which resulted in the inhibition of Sirt1 expression. Our previous study found that inhibiting CDK5 with Rosc could reverse sevoflurane-induced neuronal injury and cognitive dysfunction (Liu et al. 2017b). Increasing evidence has shown that downregulation of CDK5 prevents cognitive dysfunction and hippocampal degeneration under stress conditions (Gutierrez-Vargas et al. 2015; Yang et al. 2014). Studies have also shown that CDK5 can suppress Sirt1 expression (Zhang et al. 2018c). Our study found that overexpression of CDK5 prevented Sirt1 expression; conversely, treatment with the CDK5 inhibitor Rosc promoted Sirt1 expression.
Autophagy plays an important regulatory role after cellular stresses and can be activated by growth factors or hypoxia, nutrient deprivation, intracellular pathogens, and endoplasmic reticulum stress (Kroemer et al. 2010). Additional studies have found that activation of autophagy can prevent the neurotoxicity of general anesthetics, including the most widely used inhalation anesthetic sevoflurane (Komita et al. 2013). In our study, we found that preventing CDK5 activation promoted autophagy after sevoflurane exposure, and that Sirt1 has an important effect on autophagy regulation. The results further illustrate that suppressing CDK5 activation promotes autophagy and downregulating Sirt1 prevents autophagy. We also discovered that autophagy exerts a protective effect on nerve cells. In vivo experiments confirmed that pretreatment with autophagy inhibitor 3-Ma reversed the Rosc protective effect against neuronal apoptosis and cognitive disorder after expose to sevoflurane, suggesting that nerve cells were sensitized to sevoflurane after inhibition of autophagy. Previous studies have established that Sirt1-dependent deacetylation of LC3 has an important function in activating autophagy (Hong et al. 2018; Qiu et al. 2018). However, the specific mechanism underlying Sirt1 regulation of autophagy should be confirmed by further studies.
Taken together, our study demonstrated that suppressing CDK5 activation with Rosc treatment promoted Sirt1-induced autophagy and that sevoflurane-induced apoptosis of primary hippocampal neurons was decreased with the activation of autophagy. However, the potential role of autophagy in protecting against sevoflurane-induced neuronal injury requires further evaluation in future studies.
References
Alkire MT, Gruver R, Miller J, McReynolds JR, Hahn EL, Cahill L (2008) Neuroimaging analysis of an anesthetic gas that blocks human emotional memory. Proc Natl Acad Sci USA 105(5):1722–1727. https://doi.org/10.1073/pnas.07116511050711651105
Chang HC, Guarente L (2013) SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153(7):1448–1460. https://doi.org/10.1016/j.cell.2013.05.027S0092-8674(13)00594-1
Fan L, Chen D, Wang J, Wu Y, Li D, Yu X (2017) Sevoflurane ameliorates myocardial cell injury by inducing autophagy via the deacetylation of LC3 by SIRT1. Anal Cell Pathol (Amst) 2017:6281285. https://doi.org/10.1155/2017/6281285
Ge HW, Hu WW, Ma LL, Kong FJ (2015) Endoplasmic reticulum stress pathway mediates isoflurane-induced neuroapoptosis and cognitive impairments in aged rats. Physiol Behav 151:16–23. https://doi.org/10.1016/j.physbeh.2015.07.008S0031-9384(15)30023-8
Ghatge S, Lee J, Smith I (2003) Sevoflurane: an ideal agent for adult day-case anesthesia? Acta Anaesthesiol Scand 47(8):917–931
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221(1):3–12. https://doi.org/10.1002/path.2697
Guo X, Lv J, Lu J, Fan L, Huang X, Hu L, Wang J, Shen X (2018) Protopanaxadiol derivative DDPU improves behavior and cognitive deficit in AD mice involving regulation of both ER stress and autophagy. Neuropharmacology 130:77–91. https://doi.org/10.1016/j.neuropharm.2017.11.033
Gutierrez-Vargas JA, Munera A, Cardona-Gomez GP (2015) CDK5 knockdown prevents hippocampal degeneration and cognitive dysfunction produced by cerebral ischemia. J Cereb Blood Flow Metab 35(12):1937–1949. https://doi.org/10.1038/jcbfm.2015.150jcbfm2015150
Hong T, Ge Z, Meng R, Wang H, Zhang P, Tang S, Lu J, Gu T, Zhu D, Bi Y (2018) Erythropoietin alleviates hepatic steatosis by activating SIRT1-mediated autophagy. Biochim Biophys Acta 1863 6:595–603
Komita M, Jin H, Aoe T (2013) The effect of endoplasmic reticulum stress on neurotoxicity caused by inhaled anesthetics. Anesth Analg 117(5):1197–1204. https://doi.org/10.1213/ANE.0b013e3182a74773
Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40(2):280–293. https://doi.org/10.1016/j.molcel.2010.09.023S1097-2765(10)00751-3
Li X, Wu Z, Zhang Y, Xu Y, Han G, Zhao P (2017) Activation of autophagy contributes to sevoflurane-induced neurotoxicity in fetal rats. Front Mol Neurosci 10:432. https://doi.org/10.3389/fnmol.2017.00432
Lin D, Zuo Z (2011) Isoflurane induces hippocampal cell injury and cognitive impairments in adult rats. Neuropharmacology 61(8):1354–1359. https://doi.org/10.1016/j.neuropharm.2011.08.011S0028-3908(11)00344-3
Liu B, Xia J, Chen Y, Zhang J (2017a) Sevoflurane-induced endoplasmic reticulum stress contributes to neuroapoptosis and BACE-1 expression in the developing brain: the role of eIF2alpha. Neurotox Res 31(2):218–229. https://doi.org/10.1007/s12640-016-9671-z10.1007/s12640-016-9671-z
Liu J, Yang J, Xu Y, Guo G, Cai L, Wu H, Zhao Y, Zhang X (2017b) Roscovitine, a CDK5 inhibitor, alleviates sevoflurane-induced cognitive dysfunction via regulation tau/GSK3beta and ERK/PPARgamma/CREB signaling. Cell Physiol Biochem 44(2):423–435. https://doi.org/10.1159/000485008000485008
Lv X, Yan J, Jiang J, Zhou X, Lu Y, Jiang H (2017) MicroRNA-27a-3p suppression of peroxisome proliferator-activated receptor-gamma contributes to cognitive impairments resulting from sevoflurane treatment. J Neurochem 143(3):306–319. https://doi.org/10.1111/jnc.14208
Michan S, Sinclair D (2007) Sirtuins in mammals: insights into their biological function. Biochem J 404(1):1–13. https://doi.org/10.1042/BJ20070140
Ou X, Lee MR, Huang X, Messina-Graham S, Broxmeyer HE (2014) SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 32(5):1183–1194. https://doi.org/10.1002/stem.1641
Qiu R, Li W, Liu Y (2018) MicroRNA-204 protects H9C2 cells against hypoxia/reoxygenation-induced injury through regulating SIRT1-mediated autophagy. Biomed Pharmacother 100:15–19. https://doi.org/10.1016/j.biopha.2018.01.165
Rohan D, Buggy DJ, Crowley S, Ling FK, Gallagher H, Regan C, Moriarty DC (2005) Increased incidence of postoperative cognitive dysfunction 24 hr after minor surgery in the elderly. Can J Anaesth 52(2):137–142. https://doi.org/10.1007/BF03027718
Shen X, Dong Y, Xu Z, Wang H, Miao C, Soriano SG, Sun D, Baxter MG, Zhang Y, Xie Z (2013) Selective anesthesia-induced neuroinflammation in developing mouse brain and cognitive impairment. Anesthesiology 118(3):502–515. https://doi.org/10.1097/ALN.0b013e3182834d77
Stunkel W, Campbell RM (2011) Sirtuin 1 (SIRT1): the misunderstood HDAC. J Biomol Screen 16(10):1153–1169. https://doi.org/10.1177/10870571114221031087057111422103
Sun T, Jiao L, Wang Y, Yu Y, Ming L (2018) SIRT1 induces epithelial-mesenchymal transition by promoting autophagic degradation of E-cadherin in melanoma cells. Cell Death Dis 9(2):136. https://doi.org/10.1038/s41419-017-0167-410.1038/s41419-017-0167-4
Wang Y, Wang W, Li D, Li M, Wang P, Wen J, Liang M, Su B, Yin Y (2014) IGF-1 alleviates NMDA-induced excitotoxicity in cultured hippocampal neurons against autophagy via the NR2B/PI3K-AKT-mTOR pathway. J Cell Physiol 229(11):1618–1629. https://doi.org/10.1002/jcp.24607
Wiklund A, Granon S, Faure P, Sundman E, Changeux JP, Eriksson LI (2009) Object memory in young and aged mice after sevoflurane anaesthesia. NeuroReport 20(16):1419–1423. https://doi.org/10.1097/WNR.0b013e328330cd2b
Xiao X, Zhu Y, Bu J, Li G, Liang Z, Yang L, Hou B (2017) The autophagy inhibitor 3-methyladenine restores sevoflurane anesthesiainduced cognitive dysfunction and neurons apoptosis. Pharmazie 72(4):214–218. https://doi.org/10.1691/ph.2017.6872
Yang L, Gu X, Zhang W, Zhang J, Ma Z (2014) Cdk5 inhibitor roscovitine alleviates neuropathic pain in the dorsal root ganglia by downregulating N-methyl-D-aspartate receptor subunit 2A. Neurol Sci 35(9):1365–1371. https://doi.org/10.1007/s10072-014-1713-9
Zhang Q, Zhang P, Qi GJ, Zhang Z, He F, Lv ZX, Peng X, Cai HW, Li TX, Wang XM, Tian B (2018a) Cdk5 suppression blocks SIRT1 degradation via the ubiquitin-proteasome pathway in Parkinson's disease models. Biochim Biophys Acta 1862 6:1443–1451. https://doi.org/10.1016/j.bbagen.2018.03.021
Zhang YP, Lou Y, Hu J, Miao R, Ma F (2018b) DHA supplementation improves cognitive function via enhancing Abeta-mediated autophagy in Chinese elderly with mild cognitive impairment: a randomised placebo-controlled trial. J Neurol Neurosurg Psychiatry 89(4):382–388. https://doi.org/10.1136/jnnp-2017-316176jnnp-2017-316176
Zhang Z, Zhang P, Qi GJ, Jiao FJ, Wang QZ, Yan JG, He F, Zhang Q, Lv ZX, Peng X, Cai HW, Chen X, Sun N, Tian B (2018c) CDK5-mediated phosphorylation of Sirt2 contributes to depressive-like behavior induced by social defeat stress. Biochim Biophys Acta 1864 2:533–541. https://doi.org/10.1016/j.bbadis.2017.11.012
Zheng YL, Zhang X, Fu HX, Guo M, Shukla V, Amin ND, Jing E, Ba L, Luo HY, Li B, Lu XH, Gao YC (2016) Knockdown of expression of Cdk5 or p35 (a Cdk5 activator) results in podocyte apoptosis. PLoS ONE 11(8):e0160252. https://doi.org/10.1371/journal.pone.0160252PONE-D-15-43733
Zhou YF, Wang QX, Zhou HY, Chen G (2016) Autophagy activation prevents sevoflurane-induced neurotoxicity in H4 human neuroglioma cells. Acta Pharmacol Sin 37(5):580–588. https://doi.org/10.1038/aps.2016.6aps20166
Acknowledgements
This study was supported Tongji Medical School, Shanghai Tongji Hospital, Tongji University.
Funding
This study was supported by the National Natural Science Foundation (No. 81600934 to Jianhui Liu), National Natural Science Foundation of China (81974155 to Jianhui Liu), Pujiang Talent Programme (2019PJD049 to Jianhui Liu), the Natural Science Foundation of Shanghai, China (No. 16ZR1432200 to Jianhui Liu), and Medicine guidance of Science and Technology Commission of Shanghai Municipality (No. 16411967700 to Jianhui Liu).
Author information
Authors and Affiliations
Contributions
XZ and JL designed and conceived the study. XY, WZ, HW, SF, and JY performed the analysis and experiments. SL and YZ drafted the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors declare that they have no competing interest.
Ethical Approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were approved by the Shanghai Tongji Hospital, Tongji Medical School, Tongji University. The experiments of this manuscript comply with the current laws of the country in which they were performed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yang, X., Zhang, W., Wu, H. et al. Downregulation of CDK5 Restores Sevoflurane-Induced Cognitive Dysfunction by Promoting SIRT1-Mediated Autophagy. Cell Mol Neurobiol 40, 955–965 (2020). https://doi.org/10.1007/s10571-020-00786-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-020-00786-6