
Abstract
Accumulation of α-synuclein (α-syn) species in dopaminergic neurons is one of the main hallmarks of Parkinson’s disease (PD). Several factors have been associated with α-syn aggregation process, including an impairment of the proper protein degradation, which might drive the neurons toward an alternative and/or additional clearance mechanism that involves the release of undigested material from the cell. It has been reported that extracellular α-syn, released by stressed and/or degenerating neurons, might widely contribute to the neuronal toxicity and degeneration. Therefore, the uptake and clearance of misfolded/aggregated proteins is a key process to control extracellular deposition of α-syn aggregates, the spreading and progression of the disease. All the main brain cell types, neurons, astrocytes and microglia are able to internalize and degrade extracellular α-syn, however, glial cells appear to be the most efficient scavengers. Accumulating evidence indicates that the endocytosis of α-syn species might be conformation-sensitive, cell- and receptor-type specific, making the scenario highly complex. In this review, we will shed light on the different endocytosis mechanisms and receptors recruited for the uptake and clearance of pathological α-syn forms with a special focus on glial cells. Moreover, we will discuss how PD-related genes, in addition to α-syn itself, may alter the endo-lysosomal pathway causing an impairment of clearance, which, in turn, lead to accumulation of toxic species, dysfunctions of glia physiology and progression of the disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
α-Synuclein and Parkinson’s Disease
α-Synuclein (α-syn) is a 140 amino acids protein abundantly expressed in the brain (Ltic et al. 2004) and predominantly localized at pre-synaptic terminals of neurons (Iwai et al. 1995; Jakes et al. 1994). Whether α-syn is also expressed in glial cells under normal condition is somewhat still controversial; it would appear that, if it exists, it is expressed at very low levels (Mori et al. 2002). To date α-syn physiological functions have not been conclusively established, however, numerous studies associate α-syn to synaptic vesicles biogenesis and dynamics and to neurotransmission (Stefanis 2012). On the other hand, pathologically, it is well known that α-syn plays a crucial role in the pathogenesis of PD. α-Syn gene, SNCA, was the first gene to be linked to PD and is arguably one of the most important given that: (i) duplications and triplications of the gene, as well as point mutations, cause familial disease, (ii) variation at its locus is the major genetic risk factor for sporadic disease and (iii) the protein is the principal constituent of Lewy bodies (LB) and neurites (LN) (Devine et al. 2011). LB and LN in the surviving neurons, together with the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), represent the principal characteristics of the disease (Lees et al. 2009; Spillantini et al. 1997). Of interest, LB and LN are mainly composed of aggregated α-syn (Goedert et al. 2017), thus suggesting that the aggregation process of α-syn and its subsequent accumulation in amyloid deposits is a crucial event in the pathogenesis of both sporadic and familial PD.
α-Syn is a natively unfolded and soluble protein with a conserved amphipathic N-terminus, an acidic C-terminus and a hydrophobic central domain responsible of both oligomerization and fibrillization processes (Giasson et al. 2001). Several factors have been reported to modulate α-syn aggregation and amyloid fibrils formation. Besides duplication and triplication of the locus, even missense mutations (A53T, A30P, E46K and the recently discovered H50Q) render α-syn protein more prone to misfolding and aggregation (Greenbaum et al. 2005; Conway et al. 1998; Ghosh et al. 2013). Moreover, α-syn aggregation would depend on posttranslational modifications (serine 129 phosphorylation) (Fujiwara et al. 2002), interaction with other proteins and/or biomolecules (Recchia et al. 2004) and the presence of specific chemical parameters such as acidic pH (Buell et al. 2014). Furthermore, intraneuronal accumulation of α-syn could result also by an impairment of the proper protein degradation, which might drive the system toward an alternative and/or additional clearance mechanism that involve the release of undigested material from the cell (Lopes da Fonseca et al. 2015). In this regard, several studies have demonstrated that α-syn toxic species can be released by stressed and/or degenerating neurons and contribute to the spreading of the pathology (Bieri et al. 2018; Lopes da Fonseca et al. 2015). Intriguingly, accumulating evidence highlights that glial cells can uptake and clear extracellular α-syn aggregates (Loria et al. 2017; Sacino et al. 2017), and this suggests that even dysfunctions of non-neuronal cells might lead to accumulation of extracellular aggregates, spreading and progression of the disease.
Glia: Functions and Dysfunctions
Three different types of cells compose the glial cell family, microglia, astrocytes and oligodendrocytes with each type characterized by specific functions. While oligodendrocytes are specialized in generating and sustaining axon myelination in the Central Nervous System (CNS) (Baumann and Pham-Dinh 2001) microglia and astrocytes are more skilled in maintaining brain microenvironment homeostasis (Joe et al. 2018), and this makes them as main actors during the pathology. In this paragraph, we take a closer look at some physiological functions and dysfunctions of astrocytes and microglial cells in relation to PD.
Microglial cells, the resident phagocytes of the brain, perform essential roles in the normal physiology of the CNS, including monitoring synapses, pruning synapses during synaptic development and shaping new born neurons in adult brain (Wake and Fields 2011). Of relevance, they constitute the first barrier of the innate immune response in the brain and, therefore, microglia are the key players upon an inflammatory stimulus. Microglial cells constantly screen the parenchyma with their vastly branched and motile processes, ready to detect any changes in the surrounding tissue (Nimmerjahn et al. 2005). Upon detection of abnormal changes, such as dead neurons or misfolded/aggregated proteins, they switch from a resting and ramified phenotype into an ameboid, activated phenotype to initiate a repair program through the release of inflammatory mediators aimed to remove foreigner materials by phagocytosis (Garden and Möller 2006). Although a well-regulated inflammatory process is essential for tissue repair and CNS integrity, an excessive and protracted microglia activation and inflammation can turn cytotoxic, leading to significant cellular and tissue damage that promotes the progression of different diseases including PD. In this regard, several animal models of PD reported activated microglial cells, inflammation and depletion of dopaminergic neurons, highlighting the neurotoxic impact of microglia in PD (Członkowska et al. 1996; Mosley et al. 2006; Joers et al. 2017). During the pathology, activated microglia might induce neuronal injury and death through production of different factors, including pro-inflammatory cytokines, reactive oxygen species and glutamate (Mosley et al. 2006). Among reactive oxygen species, microglial cells produce nitric oxide (NO) and superoxide, which are responsible for the massive oxidative stress affecting dopaminergic neurons viability (Appel et al. 2010). Moreover, microglia propagate the inflammatory response by presentation of antigen-derived peptides through MHC II to CD4 T lymphocytes, which process has been reported to contribute to dopaminergic neurons degeneration (Harms et al. 2013). Of note, elevated levels of pro-inflammatory cytokines as well as HLA-positive reactive microglia have been observed in the vicinity of dopaminergic neurons also in the SNpc of PD postmortem brains (Mogi et al. 1994; McGeer et al. 1988), supporting the notion that over-activated microglia widely contribute to neurodegeneration in PD.
Astrocytes, instead, the most populous glial subtype, are responsible for a wide variety of complex and essential functions implicated in preserving the integrity of the brain. Astrocytic cells primarily support neuronal activity and function by providing important growth factors, energy generated by glucose taken up from blood and glutamine that then neurons convert in glutamate for neurotransmission. Moreover, they tightly control the external chemical microenvironment, specifically potassium ions concentration, extracellular glutamate and water content (Joe et al. 2018). Although few studies have investigated the roles of astrocytes under pathological conditions, recent evidence highlighted the impact and the importance of the pathophysiology of these cells during the disease (Booth et al. 2017). They are responsible of the uptake of the excessive glutamate in the synaptic cleft to prevent receptor over-activation and excitotoxicity, a trademark of many neurodegenerative diseases comprising PD (Kim et al. 2011; Murphy-Royal et al. 2017). Furthermore, they participate with microglia to the neuroinflammation by releasing pro-inflammatory mediators. In this regard, Lee and colleagues showed that astrocytes exposed to neuron-derived α-syn aggregates underwent changes in the gene expression profile that reflect the inflammatory response with induction of different pro-inflammatory cytokines and chemokines (Lee et al. 2010). Moreover, Rannikko et al. (2015) reported that primary astrocytes exposed to α-syn display activation of TLR4 pathway with production of IL-1β, COX-2 and TNF-α cytokines, supporting the idea that astrocytes actively contribute to the inflammation. Finally, even if not recognized as specialized phagocytic cells, astrocytes contribute to the uptake and clearance of died neurons and disease-specific protein aggregates (Jung and Chung 2018; Booth et al. 2017).
Overall, these observations indicate that loss or alterations of glial functions may be directly associated with pathological mechanisms underlying PD. In the next paragraphs, we will focus on the ability of glial cells to endocyte and clear α-syn. Specifically, we will shed light on the different mechanisms and receptors recruited to internalize extracellular α-syn species and on how PD-related genes, in addition to α-syn itself, may alter the endo-lysosomal pathway causing an impairment of clearance, which, in turn, might lead to accumulation of toxic species, dysfunctions of glia physiology and spreading of the disease.
Endocytosis and Clearance of α-syn Species: A Glial Point of View
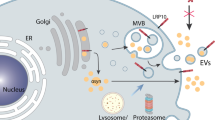
Endocytosis is the process by which a cell internalizes membrane proteins, ligands, soluble molecules and particles from the extracellular milieu into the cell by endocytic vesicles (Lim et al. 2011). Endocytosis could be distinguished in two categories, clathrin-independent and clathrin-dependent endocytosis. The clathrin-independent pathway comprehends two different types of endocytosis, the large-scale pathways that include the macropinocytosis and the phagocytosis, and the small-scale pathways further divided in dynamin-dependent or independent mechanism. Through phagocytosis, a cell is able to internalize particulates bigger than 0.5 µm within a plasma membrane envelope, while by micropinocytosis a cell uptakes large quantities of solute or soluble ligands (Gordon 2016). Among the small-scale clathrin-independent endocytosis, which leads to the internalization of particles smaller than 200 nm, the most studied and best characterized process is the caveolar endocytic system, a dynamin-dependent pathway, mediated by the integral membrane proteins caveolins and a complex of peripheral membrane proteins, the cavins (Parton et al. 2013). While, through the clathrin-dependent endocytosis the cell uptakes molecules from the cell membrane through clathrin-coated vesicles. Different cargoes can be packaged by this pathway using a range of specific adaptor proteins, such as adaptor protein 2 (AP2) that mediates on one hand interaction with clathrin and, on the other hand, with other accessory proteins linked to specific cargoes (McMahon and Boucrot 2011). When cargo has been taken up, it is sorted in early endosomes targeted to mature endosomes and then to lysosomes for degradation or sent back to membrane for recycling (Grant and Juile 2009) (Fig. 1a).

a Schematic representation of the receptors recruited for internalization of α-syn species by glial cells. TLR2 and TLR4 receptors mediate α-syn species endocytosis through clathrin-dependent pathway, while the ganglioside GM1, Fcγ and scavenger receptors (SRs) have been reported to mediate α-syn species phagocytosis through clathrin-independent process. The internalization of material drives to formation of an early endosome (EE) that then matures to late endosome (LE) and fuses with lysosome (L), which undergoes a progressive acidification leading to α-syn species degradation. b PD-related genes might lead to an impairment of α-syn aggregates degradation and accumulation of toxic species. LRRK2 pathological mutants through physiological interaction with Rab proteins might slow down the fusion of endosomes and thus the functionality of the endocytic pathway. Mutations in ATP13A2 and GBA might compromise lysosomal structure and activity, causing accumulation of undigested α-syn species, which, with time, might lead to glial physiology dysfunctions, spreading and progression of the disease
All the major brain cell types (neurons, astrocytes and microglia) have endocytic activity and are capable of uptaking extracellular α-syn aggregates, however, to date microglia appear to be the most efficient scavengers (Lee et al. 2008a). Although several studies reported α-syn aggregates internalized in microglial cells, the precise process underlying this function is not completely understood. Park et al. demonstrated that immortalized BV2 cells uptake α-syn through the ganglioside GM1 and the mechanism seems to be clathrin-, dynamin- and caveolae-independent, but linked to lipid rafts of plasma membrane (Park et al. 2009). Other studies reported that phagocytosis of α-syn aggregated forms occurs through Fcγ or scavenger receptor (Choi et al. 2015; Cao et al. 2012; Zhang et al. 2007). In addition, different Toll-like receptors (TLRs) have been associated with α-syn pathological forms uptake both in in vitro (Kim et al. 2013; Fellner et al. 2013; Roodveldt et al. 2013) and in in vivo studies (Stefanova et al. 2011; Kim et al. 2013). Specifically, TLR4 is required for fibrillary α-syn internalization (Fellner et al. 2013), and, of interest, TLR4 deficiency impairs microglia endocytosis resulting in poor α-syn clearance and increased mediated-neurodegeneration in a multiple system atrophy model (Stefanova et al. 2011). Besides TLR4, also microglial TLR2 has been shown to bind extracellular α-syn (Kim et al. 2013). Of note, Kim and colleagues proposed that TLR-ligand activity of α-syn may be conformation-sensitive, given that only oligomeric species can interact with TLR2 (Kim et al. 2013). Supporting the idea that internalization of α-syn might depend on the conformation state of the protein, two different studies showed that microglia in the presence of α-syn monomeric form displayed an enhanced phagocytic function, which seems to be inhibited by aggregated species (Park et al. 2008; Choi et al. 2015). Interestingly, Lee et al. reported that the clearance of α-syn pathological species is significantly slowed down in activated and inflamed microglia (Lee et al. 2008a), suggesting that the phagocytic function of microglia may be impaired during the disease.
Although very few studies have investigated the endocytosis and clearance of α-syn in astrocytes, even these cells can be sweepers of α-syn aggregates. Recent literature showed that α-syn toxic species can be efficiently internalized by astrocytic cells and degraded through the lysosomal pathway (Lindström et al. 2017; Loria et al. 2017; Sacino et al. 2017). Moreover, Loria et al., by using co-cultures of primary neurons and astrocytes observed that fibrillary α-syn can be transferred to neighboring cells, and, interestingly, they showed that α-syn is more transferred from astrocytes to astrocytes and from neurons to astrocytes but less from astrocytes to neurons. Furthermore, the authors demonstrated that, differently from neurons, astrocytes are able to efficiently degrade α-syn (Loria et al. 2017). These results suggest that also astrocytes have an active role in uptaking and clearing extracellular α-syn species released by stressed and/or degenerating neurons. Intriguingly, it has been shown that upon extensive uptake of α-syn aggregates, astrocytic cells exhibit large intracellular deposits due to the overwhelming of endo-lysosomal machinery and to an incomplete digestion (Lindström et al. 2017; Sacino et al. 2017), indicative of an impaired phagocytic activity even for astrocytes under pathological conditions. In this context, in the first phases of the disease glial cells might play a beneficial role with the endocytosis and clearance of α-syn species; however, during the pathology a persistent generation of α-syn aggregates might compromise the endo-lysosomal system causing an accumulation of undigested α-syn species, which, with time, might lead to glia physiology dysfunctions, spreading and progression of the disease.
As well as conformation-dependent, evidence indicates that the uptake of extracellular α-syn might even be cell- and receptor-type specific. In this context, it has been shown that heparan sulfate, PrPC prion protein and lymphocyte-activation gene 3 (LAG3) mediate the uptake of fibrillary α-syn, but not of its soluble form, exclusively in neuronal cells and not in microglia and astrocytes (Aulić et al. 2017; Ihse et al. 2017; Mao et al. 2016). Additionally, supporting the idea of cell-type specificity, in contrast to what observed in microglia, α-syn aggregates endocytosis is not mediated through TLR4 in astrocytic cells (Rannikko et al. 2015; Fellner et al. 2013). Furthermore, a recent study by Outeiro’s group showed that in a neuron-like cell line the internalization of α-syn is mediated by its interaction with the plasma membrane (Masaracchia et al. 2018). In support of this, even Lee and colleagues observed in the same cell line that the monomeric form of α-syn might enter the cell by translocation across the plasma membrane (Lee et al. 2008b), thus removing the recruitment of any receptor for the endocytosis. Taken together, these findings indicate that the uptake/clearance of α-syn might involve different pathways and receptors and be unique for each cell type and α-syn form, making the scenario highly complex and needy of deeply investigations.
A Role of PD-Related Genes in the Clearance of α-syn Pathological Species?
As discussed earlier, the clearance of extracellular α-syn involves the internalization and consequent degradation process, which occurs through the endo-lysosomal vesicles (Fig. 1a). A number of genes linked to PD, either carrying causal mutations (LRRK2 and ATP13A2) or more common variants acting as risk factors (heterozygous mutations in glucocerebrosidase, GBA), have been reported to play a role in the endo-lysosomal process. Accumulating literature provided strong evidence that LRRK2 directly impacts the endocytic machinery through interaction with Rab membrane-associated small GTPases, which control the maturation and fusion of a subset of endosomes that lead to degradation of lysosomal content (Roosen and Mark 2016). Specifically, Rab5 (Heo et al. 2010), Rab7L1 (Beilina et al. 2014), Rab32 and Rab38 (Waschbüsch et al. 2014) interact with LRRK2, and Rab3a/b/c, Rab8a/b, Rab10, Rab12, Rab35 and Rab43 have been reported to be direct physiological substrates of LRRK2 kinase activity (Steger et al. 2016, 2017; Bae et al. 2018; Yu et al. 2018). Additionally, it has been reported that LRRK2 with pathological mutations results in an increased phosphorylation (Steger et al. 2016) and physiological function of Rab proteins (Beilina et al. 2014). Moreover, supporting the notion that LRRK2 regulates the endo-lysosomal pathway, LRRK2 has been reported to localize on lysosomes under control (Alegre-Abarrategui et al. 2009) and stress conditions (Eguchi et al. 2018) and positively regulate the clearance activity in microglia (Schapansky et al. 2014). Interestingly, Melrose’s group demonstrated that microglial cells with LRRK2 deficiency exhibited an enhanced endocytosis of monomeric α-syn compared with wild-type cells likely due to an increase in Rab5-positive endosomes (Maekawa et al. 2016). Altogether, these findings indicate that LRRK2 controls diverse vesicular trafficking events of the endo-lysosomal pathway and suggest that LRRK2 mutations might widely impact the endocytic machinery and the digestion of unfolded/aggregated proteins in glial cells.
GBA and ATP13A2 genes, a risk factor and a causal mutation of PD, respectively, encode proteins related to lysosome biology and functionality. Specifically, glucocerebrosidase (GCase) is an enzyme associated with lysosomal membranes where it converts glucosyl-ceramides into ceramide. Of interest, it has been reported that GCase activity modulates α-syn levels (Manning-Boğ et al. 2009). In this regard, Mazzulli et al. showed that functional loss of GCase impacts lysosomal protein degradation causing aggregation of α-syn and neurotoxicity mediated by aggregates-dependent mechanisms (Mazzulli et al. 2011). In addition to a loss of function, also a GCase gain of function has been reported to compromise lysosomal activity leading to accumulation of α-syn in neuronal cells (Cullen et al. 2011). Even mutations in ATP13A2, a transmembrane lysosomal P5-type ATPase, have been reported to cause lysosomal degradation dysfunctions likely due to an impaired targeting of the ATPase to lysosomes (Podhajska et al. 2012; Ugolino et al. 2011). In this context, studies performed in dopaminergic cells reveal that ATP13A2 deficiency induces instability of the lysosomal membrane, impaired vesicle acidification and decreased degradation of lysosomal substrates (Dehay et al. 2012a, b; Usenovic et al. 2012). Interestingly, as well as GCase, cells with ATP13A2 mutants or ATP13A2 deficiency displayed a marked accumulation of α-syn due to an impairment of lysosomal proteolysis (Dehay et al. 2012b; Usenovic et al. 2012), indicating that the proper physiology of lysosomes is crucial for the quality control system and the cell physiology. Overall, although different studies showed a link between PD-related genes and lysosomal functionality in neuronal cells, whether these genes impact endo-lysosomal system in astrocytes and microglia is still unknown and needy of investigations. In support of a role of ATP13A2 on lysosomal activity of glial cells, Qiao et al. found that astrocytes expressed high levels of ATP13A2 and its deficiency in astrocytes induces lysosomal permeabilization, which leads to cathepsin B release and cell activation (Qiao et al. 2016).
Taken together, these observations propose that, in addition to α-syn itself, mutations in other PD-related genes might compromise the clearance through alterations of vesicular trafficking events of the endo-lysosomal pathway (Fig. 1b). The uptake and clearance of α-syn toxic species is a key event to control the extracellular deposition of α-syn aggregates and alterations of this process might cause accumulation of toxic species, which, with time, might lead to dysfunctions of glia physiology, spreading and progression of PD.
Abbreviations
- α-syn:
-
α-Synuclein
- PD:
-
Parkinson’s disease
- LB:
-
Lewy bodies
- LN:
-
Lewy neurites
- CNS:
-
Central nervous system
- Snpc:
-
Substantia nigra pars compacta
- AP2:
-
Adaptor protein 2
- TLR:
-
Toll-like receptors
- LAG3:
-
Lymphocyte-activation gene 3
- GCase:
-
Glucocerebrosidase GCase
- pffs:
-
pre-formed fibrils
References
Alegre-Abarrategui J, Christian H, Michele MP, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins RR (2009) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum Mol Genet 18(21):4022–4034. https://doi.org/10.1093/hmg/ddp346
Appel SH, Beers DR, Henkel JS (2010) T cell-microglial dialogue in Parkinson’s disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol 31(1):7–17. https://doi.org/10.1016/j.it.2009.09.003
Aulić S, Masperone L, Narkiewicz J, Isopi E, Bistaffa E, Ambrosetti E, Pastore B et al (2017) α-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci Rep 7(1):10050. https://doi.org/10.1038/s41598-017-10236-x
Bae E-J, Kim D-K, Kim C, Mante M, Adame A, Rockenstein E, Ulusoy A et al (2018) LRRK2 kinase regulates α-Synuclein propagation via RAB35 phosphorylation. Nat Commun 9(1):3465. https://doi.org/10.1038/s41467-018-05958-z
Baumann N, Pham-Dinh D (2001) Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 81(2):871–927. https://doi.org/10.1152/physrev.2001.81.2.871
Beilina A, Rudenko IN, Kaganovich A, Civiero L, Chau H, Suneil K, Kalia LV, Kalia et al (2014) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci USA 111(7):2626–2631. https://doi.org/10.1073/pnas.1318306111
Bieri G, Gitler AD, Brahic M (2018) Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol Dis 109:219–225. https://doi.org/10.1016/j.nbd.2017.03.007
Booth HDE, Hirst WD, Wade-Martins R (2017) The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci 40(6):358–370. https://doi.org/10.1016/j.tins.2017.04.001
Buell AK, Galvagnion C, Gaspar R, Sparr E, Vendruscolo M, Knowles TP, Linse S, Dobson CM (2014) Solution conditions determine the relative importance of nucleation and growth processes in α-Synuclein aggregation. Proc Natl Acad Sci USA 111(21):7671–7676. https://doi.org/10.1073/pnas.1315346111
Cao S, Standaert DG, Ashley SH (2012) The gamma chain subunit of Fc receptors is required for alpha-Synuclein-induced pro-inflammatory signaling in microglia. J Neuroinflamm 9(1):259. https://doi.org/10.1186/1742-2094-9-259
Choi YR, Kang S-J, Kim J-M, Lee S-J, Jou I, Joe E-H, Park SM (2015) FcγRIIB mediates the inhibitory effect of aggregated α-Synuclein on microglial phagocytosis. Neurobiol Dis 83:90–99. https://doi.org/10.1016/j.nbd.2015.08.025
Conway KA, Harper JD, Lansbury PT (1998) Accelerated in vitro fibril formation by a mutant α-Synuclein linked to early-onset Parkinson disease. Nat Med 4(11):1318–1320. https://doi.org/10.1038/3311
Cullen V, Sardi SP, Ng J, Xu Y-H, Sun Y, Tomlinson JJ, Kolodziej P et al (2011) Acid β-glucosidase mutants linked to gaucher disease, Parkinson disease, and lewy body dementia alter α-Synuclein processing. Ann Neurol 69(6):940–953. https://doi.org/10.1002/ana.22400
Członkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Członkowski A (1996) Microglial reaction in MPTP (1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine) Induced Parkinson’s disease mice model. Neurodegeneration 5(2):137–143
Dehay B, Martinez-Vicente M, Ramirez A, Perier C, Klein C, Vila M, Bezard E (2012a) Lysosomal dysfunction in Parkinson disease: ATP13A2 gets into the groove. Autophagy 8(9):1389–1391. https://doi.org/10.4161/auto.21011
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron M-H, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E (2012b) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci USA 109(24):9611–9616. https://doi.org/10.1073/pnas.1112368109
Devine MJ, Gwinn K, Singleton A, Hardy J (2011) Parkinson’s disease and α-Synuclein expression. Mov Disord 26(12):2160–2168. https://doi.org/10.1002/mds.23948
Eguchi T, Kuwahara T, Sakurai M, Komori T, Fujimoto T, Ito G, Yoshimura S-I et al (2018) LRRK2 and its substrate rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc Natl Acad Sci USA 115(39):E9115–E9124. https://doi.org/10.1073/pnas.1812196115
Fellner L, Irschick R, Schanda K, Reindl M, Klimaschewski L, Poewe W, Wenning GK, Stefanova N (2013) Toll-like receptor 4 is required for α-Synuclein dependent activation of microglia and astroglia. Glia 61(3):349–360. https://doi.org/10.1002/glia.22437
Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002) Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4(2):160–164. https://doi.org/10.1038/ncb748
Garden GA, Möller T (2006) Microglia biology in health and disease. J Neuroimmune Pharmacol 1(2):127–137. https://doi.org/10.1007/s11481-006-9015-5
Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, Ghosh S, Jha NN, Kumar A, Samir KM (2013) The Parkinson’s disease-associated H50Q mutation accelerates α-Synuclein aggregation in vitro. Biochemistry 52(40):6925–6927. https://doi.org/10.1021/bi400999d
Giasson BI, Murray IV, Trojanowski JQ, Lee VM (2001) A hydrophobic stretch of 12 amino acid residues in the middle of Alpha-Synuclein is essential for filament assembly. J Biol Chem 276(4):2380–2386. https://doi.org/10.1074/jbc.M008919200
Goedert M, Jakes R, Maria GS (2017) The synucleinopathies: twenty years on. J Parkinson’s Dis 7(s1):S53–S71. https://doi.org/10.3233/JPD-179005
Gordon S (2016) Phagocytosis: an immunobiologic process. Immunity 44(3):463–475. https://doi.org/10.1016/j.immuni.2016.02.026
Grant BD, Julie GD (2009) Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10(9):597–608. https://doi.org/10.1038/nrm2755
Greenbaum EA, Charles L, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Benoit IG (2005) The E46K mutation in Alpha-Synuclein increases amyloid fibril formation. J Biol Chem 280(9):7800–7807. https://doi.org/10.1074/jbc.M411638200
Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, Cron RQ, Shacka JJ, Raman C, Standaert DG (2013) MHCII is required for -Synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci 33(23):9592–9600. https://doi.org/10.1523/JNEUROSCI.5610-12.2013
Heo HY, Kim KS, Seol W (2010) Coordinate regulation of neurite outgrowth by LRRK2 and its interactor, Rab5. Exp Neurobiol 19(2):97–105. https://doi.org/10.5607/en.2010.19.2.97
Ihse E, Yamakado H, van Wijk XM, Lawrence R, Esko JD, Masliah E (2017) Cellular internalization of Alpha-Synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep 7(1):9008. https://doi.org/10.1038/s41598-017-08720-5
Iwai A, Masliah E, Yoshimoto M, Ge N, Flanagan L, de Silva HA, Kittel A, Saitoh T (1995) The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14(2):467–475
Jakes R, Spillantini MG, Goedert M (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345(1):27–32
Joe E-H, Choi D-J, An J, Eun J-H, Jou I, Park S (2018) Astrocytes, microglia, and Parkinson’s disease. Exp Neurobiol 27(2):77–87. https://doi.org/10.5607/en.2018.27.2.77
Joers V, Tansey MG, Mulas G, Anna RC (2017) Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog Neurobiol 155(August):57–75. https://doi.org/10.1016/j.pneurobio.2016.04.006
Jung Y-J, Chung WS (2018) Phagocytic roles of glial cells in healthy and diseased brains. Biomol Therapeutics. https://doi.org/10.4062/biomolther.2017.133
Kim K, Lee S-G, Kegelman TP, Su Z-Z, Swadesh K, Das R, Dash S, Dasgupta et al (2011) Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol 226(10):2484–2493. https://doi.org/10.1002/jcp.22609
Kim C, Ho D-H, Suk J-E, You S, Michael S, Kang J, Lee SJ et al (2013) Neuron-released oligomeric α-Synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4:1562. https://doi.org/10.1038/ncomms2534
Lee H-J, Suk J-E, Bae E-J, Lee SJ (2008a) Clearance and deposition of extracellular α-Synuclein aggregates in microglia. Biochem Biophys Res Commun 372(3):423–428. https://doi.org/10.1016/j.bbrc.2008.05.045
Lee H-J, Suk J-E, Bae E-J, Lee J-H, Paik SR, Lee SJ (2008b) Assembly-dependent endocytosis and clearance of extracellular α-synuclein Int J Biochem Cell Biol 40(9):1835–1849
Lee H-J, Suk J-E, Patrick C, Bae E-J, Cho J-H, Rho S, Hwang D, Masliah E, Lee SJ (2010) Direct transfer of α-Synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285(12):9262–9272. https://doi.org/10.1074/jbc.M109.081125
Lees AJ, Hardy J, Revesz T (2009) Parkinson’s disease. Lancet 373(9680):2055–2066. https://doi.org/10.1016/S0140-6736(09)60492-X
Lim J, Phey, Paul AG (2011) Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol 89(8):836–843. https://doi.org/10.1038/icb.2011.20
Lindström V, Gustafsson G, Sanders LH, Howlett EH, Sigvardson J, Kasrayan A, Ingelsson M, Bergström J, Erlandsson A (2017) Extensive uptake of α-Synuclein oligomers in astrocytes results in sustained intracellular deposits and mitochondrial damage. Mol Cell Neurosci 82:143–156. https://doi.org/10.1016/j.mcn.2017.04.009
Lopes da Fonseca T, Villar-Piqué TA, Outeiro T (2015) The interplay between alpha-synuclein clearance and spreading. Biomolecules 5 (2): 435–471. https://doi.org/10.3390/biom5020435
Loria F, Jessica Y, Vargas L, Bousset S, Syan A, Salles R, Melki, Zurzolo C (2017) α-Synuclein transfer between neurons and astrocytes indicates that astrocytes play a role in degradation rather than in spreading. Acta Neuropathol 134(5):789–808. https://doi.org/10.1007/s00401-017-1746-2
Ltic S, Perovic M, Mladenovic A, Raicevic N, Ruzdijic S, Rakic L (2004) Alpha-Synuclein is expressed in different tissues during human fetal development. J Mol Neurosci 22(3):199–204
Maekawa T, Sasaoka T, Azuma S, Ichikawa T, Melrose HL, Farrer MJ, Obata F (2016) Leucine-rich repeat kinase 2 (LRRK2) regulates α-Synuclein clearance in microglia. BMC Neurosci 17(1):77. https://doi.org/10.1186/s12868-016-0315-2
Manning-Boğ AB, Schüle B, Langston JW (2009) Alpha-Synuclein-glucocerebrosidase interactions in pharmacological gaucher models: a biological link between gaucher disease and Parkinsonism. Neurotoxicology 30(6):1127–1132. https://doi.org/10.1016/j.neuro.2009.06.009
Mao X, Ou MT, Karuppagounder SS Xiong Y, Ge P et al (2016) Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353(6307):aah3374–aah3374. https://doi.org/10.1126/science.aah3374
Masaracchia C, Hnida M, Gerhardt E, Branco T, Stahlberg MA et al (2018) Membrane binding, internalization, and sorting of Alpha-Synuclein in the cell. Acta Neuropathol Commun 6(1):79. https://doi.org/10.1186/s40478-018-0578-1
Mazzulli JR, Xu Y-H, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and α-Synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146(1):37–52. https://doi.org/10.1016/j.cell.2011.06.001
McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38(8):1285–1291
McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12(8):517–533. https://doi.org/10.1038/nrm3151
Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T (1994) Tumor necrosis factor-alpha (TNF-Alpha) increases both in the brain and in the cerebrospinal fluid from Parkinsonian patients. Neurosci Lett 165(1–2):208–210
Mori F, Tanji K, Yoshimoto M, Takahashi H, Wakabayashi K (2002) Immunohistochemical comparison of alpha- and beta-Synuclein in adult rat central nervous system. Brain Res 941(1–2):118–126
Mosley R, Lee EJ, Benner I, Kadiu M, Thomas MD, Boska K, Hasan C, Laurie, Howard EG (2006) Neuroinflammation, oxidative stress and the pathogenesis of Parkinson’s disease. Clin Neurosci Res 6(5):261–281. https://doi.org/10.1016/j.cnr.2006.09.006
Murphy-Royal C, Dupuis J, Groc L, Oliet SH (2017) Astroglial glutamate transporters in the brain: regulating neurotransmitter homeostasis and synaptic transmission. J Neurosci Res 95(11):2140–2151. https://doi.org/10.1002/jnr.24029
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318. https://doi.org/10.1126/science.1110647
Park J-Y, Paik SR, Jou I, Sang Myun Park (2008) Microglial phagocytosis is enhanced by monomeric Alpha-Synuclein, not aggregated Alpha-Synuclein: implications for Parkinson’s disease. Glia 56(11):1215–1223. https://doi.org/10.1002/glia.20691
Park J-Y, Kim KS, Lee S-B, Ryu J-S, Chung KC, Choo Y-K, Jou I, Kim J, Sang MP (2009) On the mechanism of internalization of Alpha-Synuclein into microglia: roles of ganglioside GM1 and lipid raft. J Neurochem 110(1):400–411. https://doi.org/10.1111/j.1471-4159.2009.06150.x
Parton RG, Miguel A, del Pozo (2013) Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol 14(2):98–112. https://doi.org/10.1038/nrm3512
Podhajska A, Musso A, Trancikova A, Stafa K, Moser R, Sonnay S, Glauser L, Darren JM (2012) Common pathogenic effects of missense mutations in the P-Type ATPase ATP13A2 (PARK9) associated with early-onset Parkinsonism. PLoS ONE 7(6): e39942. https://doi.org/10.1371/journal.pone.0039942
Qiao C, Yin N, Gu H-Y, Zhu J-L, Ding J-H, Lu M, Hu G (2016) Atp13a2 deficiency aggravates astrocyte-mediated neuroinflammation via NLRP3 inflammasome activation. CNS Neurosci Ther 22(6):451–460. https://doi.org/10.1111/cns.12514
Rannikko EH, Stephanie S, Weber, Philipp JK (2015) Exogenous α-Synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci 16(1):57. https://doi.org/10.1186/s12868-015-0192-0
Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P (2004) Alpha-synuclein and Parkinson’s disease. FASEB J 18(6):617–626. https://doi.org/10.1096/fj.03-0338rev
Roodveldt C, ALabrador-Garrido A, Lachaud CC, Guilliams T, Fernandez-Montesinos R, Benitez-Rondan A et al (2013) Preconditioning of microglia by α-synuclein strongly affects the response induced by toll-like receptor (TLR) stimulation. PLoS ONE 8(11):e79160. https://doi.org/10.1371/journal.pone.0079160
Roosen DA, Mark RC (2016) LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener 11(1):73. https://doi.org/10.1186/s13024-016-0140-1
Sacino AN, Mieu M, Brooks P, Chakrabarty K, Saha H, Khoshbouei TE, Golde, Benoit IG (2017) Proteolysis of α-Synuclein fibrils in the lysosomal pathway limits induction of inclusion pathology. J Neurochem 140(4):662–678. https://doi.org/10.1111/jnc.13743
Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ (2014) Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet 23(16):4201–4214. https://doi.org/10.1093/hmg/ddu138
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in lewy bodies. Nature 388(6645):839–840. https://doi.org/10.1038/42166
Stefanis L (2012) α-Synuclein in Parkinson’s disease. Cold Spring Harbor Perspect Med 2(2):a009399. https://doi.org/10.1101/cshperspect.a009399
Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Gregor KW (2011) Toll-like receptor 4 promotes α-Synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol 179(2):954–963. https://doi.org/10.1016/j.ajpath.2011.04.013
Steger M, Tonelli F, Ito G, Davies P, Trost M, Vetter M, Wachter S et al (2016) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. ELife. https://doi.org/10.7554/eLife.12813
Steger M, Diez F, Dhekne HS, Lis P, Nirujogi RS, Karayel O, Tonelli F et al (2017) Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. ELife. https://doi.org/10.7554/eLife.31012
Ugolino J, Fang S, Kubisch C, Mervyn JM (2011) Mutant Atp13a2 proteins involved in Parkinsonism are degraded by ER-associated degradation and sensitize cells to ER-stress induced cell death. Hum Mol Genet 20(18):3565–3577. https://doi.org/10.1093/hmg/ddr274
Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D (2012) Deficiency of ATP13A2 leads to lysosomal dysfunction, α-Synuclein accumulation, and neurotoxicity. J Neurosci 32(12):4240–4246. https://doi.org/10.1523/JNEUROSCI.5575-11.2012
Wake H, Fields RD (2011) Physiological function of microglia. Neuron Glia Biol 7(1):1–3. https://doi.org/10.1017/S1740925X12000166
Waschbüsch D, Michels H, Strassheim S, Ossendorf E, Kessler D, Gloeckner CJ, Barnekow A (2014) LRRK2 transport is regulated by its novel interacting partner Rab32. PLoS ONE 9(10):e111632. https://doi.org/10.1371/journal.pone.0111632
Yu M, Arshad M, Wang W, Zhao D, Xu L, Zhou L (2018) LRRK2 mediated Rab8a phosphorylation promotes lipid storage. Lipids Health Dis 17(1):34. https://doi.org/10.1186/s12944-018-0684-x
Zhang W, Dallas S, Zhang D, Guo J-P, Pang H, Wilson B, Miller DS et al (2007) Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T Mutant Alpha-synuclein. Glia 55(11):1178–1188. https://doi.org/10.1002/glia.20532
Acknowledgements
We are grateful to the financial support of CARIPLO Foundation (Grant No. 2016-0428).
Author information
Authors and Affiliations
Contributions
AF and IR conceived the paper; AF, MG and IR wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest for this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Filippini, A., Gennarelli, M. & Russo, I. α-Synuclein and Glia in Parkinson’s Disease: A Beneficial or a Detrimental Duet for the Endo-Lysosomal System?. Cell Mol Neurobiol 39, 161–168 (2019). https://doi.org/10.1007/s10571-019-00649-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-019-00649-9