
Abstract
Hemorrhagic stroke is a devastating clinical event with no effective medical treatment. Neuroinflammation, which follows a hemorrhagic stroke, is an important element that involves both acute brain injury and subsequent brain rehabilitation. Therefore, delineating the key inflammatory mediators and deciphering their pathophysiological roles in hemorrhagic strokes is of great importance in the development of novel therapeutic targets for this disease. The NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is a multi-protein complex that is localized within the cytoplasm. This NOD-like receptor orchestrates innate immune responses to pathogenic organisms and cell stress through the activation of caspase-1 and the maturation of the proinflammatory cytokines such as interleukin-1β (IL-1β) and IL-18. Mounting evidence has demonstrated that when the NLRP3 inflammasome is activated, it exerts harmful effects on brain tissue after a hemorrhagic stroke. This review article summarizes the current knowledge regarding the role and the underlying mechanisms of the NLRP3 inflammasome in the pathophysiological processes of hemorrhagic strokes. A better understanding of the function and regulation of the NLRP3 inflammasome in hemorrhagic strokes will provide clues for devising novel therapeutic strategies to fight this disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
There are two types of hemorrhagic strokes: the intracerebral hemorrhage (ICH) and the subarachnoid hemorrhage (SAH). Hemorrhagic stroke is a devastating disease that accounts for 10–20% of all strokes (Xi et al. 2014; Rinkel and Algra 2011). The pathophysiology of hemorrhagic strokes is complicated and involves multiple pathogenic mechanisms (e.g., inflammation, oxidative stress, and apoptosis) (Xi et al. 2006; Li et al. 2014). Inflammation is one of the key elements in the pathophysiological process of hemorrhagic stroke (Zhou et al. 2014; Caner et al. 2012). The inflammatory responses to hemorrhagic stroke overlap with those following ischemic stroke (Wang and Dore 2007). However, important differences exist between them (Xi et al. 2006; Kleinig and Vink 2009). For instance, blood components, including inflammatory cells, chemokines, cytokines, and proteases, are introduced directly into the brain after the onset of a cerebral hemorrhage. Thrombin, which has been proven to be pivotal in the initiation of inflammation, causes intracerebral leucocyte infiltration and brain edema following a cerebral hemorrhage (Hua et al. 2007). Iron-containing compounds released by hemolysis, which are released from approximately 48 h onward, are potent inflammatory stimuli that follow a hemorrhagic stroke (Xi et al. 1998). Additionally, cerebral ischemia, which is associated with an ICH stroke, is local and irreversible and has no salvageable perihematomal penumbra (Qureshi et al. 1999). Thus, it appears to be favorable to discuss the role of inflammation in ischemic and hemorrhagic strokes. Better knowledge of the roles and regulations of these cellular events following hemorrhagic strokes is necessary for the development of novel therapeutic targets for this disease.
Inflammatory responses are involved both in acute brain injury and in the subsequent brain rehabilitation following a hemorrhagic stroke. On the one hand, excessive, inappropriate, or uncontrolled inflammatory responses amplify cellular damage within the injured brain parenchyma after the hemorrhagic stroke (Wang et al. 2013b, 2014). On the other hand, inflammatory responses act as defense mechanisms that contribute to tissue reconstruction and repair by removing dead cells and debris (Schallner et al. 2015; Zhao et al. 2007). Accordingly, maintaining the activity of neuroinflammation at an appropriate level following hemorrhagic strokes is required for the therapy that follows.
The NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome is a multi-protein complex that is distributed in the cytosol (Tschopp and Schroder 2010). This receptor, which is composed of the sensor protein NLRP3, the adapter protein apoptosis-associated speck-like protein (ASC), and the effector protein pro-caspase-1, orchestrates innate immune responses against infection and cell stress by mediating caspase-1-dependent processing and releasing proinflammatory cytokines interleukin-1β (IL-1β) and IL-18 (Leemans et al. 2011). Most importantly, the activation of the NLRP3 inflammasome is associated with the onset and progression of various diseases, such as inflammatory bowel disease, metabolic disorders, and atherosclerosis (Dashdorj et al. 2013; Yan et al. 2013; Xiao et al. 2013). In the central nervous system (CNS), the inappropriate activation of the NLRP3 inflammasome participates in the pathogenesis of both acute and chronic neurodegenerative conditions (Ito et al. 2015; Chen et al. 2015). Particularly, the accumulated evidence demonstrates that the NLRP3 inflammasome is activated and exerts detrimental effects on the brain after a hemorrhagic stroke (Chen et al. 2013; Ma et al. 2014). In this article, we summarize the current knowledge on the pathophysiological roles and regulatory mechanisms of the NLRP3 inflammasome in the pathogenesis of hemorrhagic strokes, thus providing possible clues for devising novel therapeutic strategies to treat this disease.
NLRP3 Inflammasome: Assembly and Activation
Inflammasomes are intracellular oligomeric multi-protein complexes that play critical roles in eliciting innate immune responses against microbial and damage-associated signals (Franchi et al. 2009). Canonical inflammasomes consist of a sensor, an adaptor, and an effector. Depending on the presence of different sensory molecules within such complexes, inflammasomes are broadly divided into two major categories, namely the nucleotide-binding domain leucine-rich repeat (NLR) containing family members (i.e., NLRP1, NLRP3, and NLRC4) and the PYHIN family members (i.e., AIM2 and IFI16) (Lamkanfi and Dixit 2009). Among them, the NLRP3 inflammasome is the most studied member, and it comprises the NLRP3 scaffold, the ASC adaptor, and the caspase-1 effector (Zaki et al. 2011). The NLRP3 protein contains an N-terminal pyrin domain (PYD), a central nucleotide-binding and oligomerization domain (NACHT), and a C-terminal leucine-rich repeat (LRR) domain (Bryant and Fitzgerald 2009). The LRR domains are believed to function in ligand sensing and to exert an autoregulatory role in the activation of NLRP3 (Mariathasan and Monack 2007). After sensing danger signals, the NLRP3 monomers can be activated and oligomerized to form defined oligomers (Lechtenberg et al. 2014). These ring-like structures recruit ASC monomers to induce the formation of ASC filaments or specks through homophilic PYD–PYD interactions (Lu et al. 2014). The ASC filaments/specks then recruit the cysteine proteases pro-caspase-1 to form active inflammasome complexes via the caspase recruitment domain (CARD) interaction (Proell et al. 2013). As a result, the activation of the NLRP3 inflammasome causes an autocatalytic cleavage of pro-caspase-1, leading to the processing and secretion of the proinflammatory cytokines IL-1β and IL-18 and, under certain conditions, inducing an inflammatory and lytic form of programmed cell death termed “pyroptosis” (Brydges et al. 2013; Wree et al. 2014).
Of note, the activation of the NLRP3 inflammasome is tightly regulated at the transcriptional and post-translational levels and requires two-step signals (Latz et al. 2013). The priming signal ensures adequate gene/protein expression of NLRP3, pro-IL 1β, and pro-IL 18 for efficient inflammasome formation. And the transcription of NLRP3 inflammasome components (i.e., NLRP3) can be facilitated by the activation of the Toll-like receptor (TLR)/nuclear factor kappa B (NF-κB) signaling pathway (Segovia et al. 2012). The second signal, which is conveyed by a number of stimuli including pathogen-associated molecular pattern molecules (PAMPs) and damage-associated molecular pattern molecules (DAMPs), is necessary for the assembly and activation of the NLRP3 inflammasome (Lamkanfi 2011). As summarized in other reviews, it has been proposed that three major upstream mechanisms mediate the activation of the NLRP3 inflammasome, including potassium (K+) efflux, mitochondrial reactive oxygen species (ROS) generation, and lysosomal destabilization and release of lysosomal cathepsins (Jo et al. 2016; Munoz-Planillo et al. 2013). However, the definitive molecular mechanisms for NLRP3 inflammasome activation remain elusive and require further investigation.
The Role and Regulatory Mechanisms of NLRP3 Inflammasome in a Hemorrhagic Stroke
In the CNS, mounting evidence has demonstrated that the NLRP3 inflammasome acts as a key mediator of neuroinflammation and has a prominent role in the pathogenesis and progression of strokes (Fann et al. 2013). As an example, the NLRP3 inflammasome has been proven to be important in detecting cellular damage and mediating inflammatory responses to tissue injury during an ischemic stroke (Fann et al. 2014; Ye et al. 2017a). Yang et al. found that an NLRP3 deficiency blocks blood–brain barrier (BBB) disruption, ameliorates neurovascular damage, and improves functional outcomes after an ischemic stroke in a mouse model of middle cerebral artery occlusion (MCAO) (Yang et al. 2014). Bruton’s tyrosine kinase (BTK), a member of the Tec family, can positively regulate the activation of the NLRP3 inflammasome via physically interacting with NLRP3 and ASC, thus exacerbating neurovascular damage following an ischemic stroke (Ito et al. 2015). Furthermore, the inhibition of BTK by pharmacological or genetic means severely impairs the activation of the NLRP3 inflammasome and attenuates cerebral ischemia-induced brain injury (Ito et al. 2015).
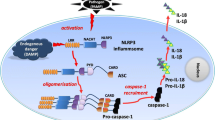
Interestingly, preclinical studies have revealed that the NLRP3 inflammasome plays a critical role in regulating neuroinflammation after a hemorrhagic stroke (Table 1). In the following sections, we will review the research literature on the role of the NLRP3 inflammasome in experimental models of hemorrhagic stroke and survey the underlying molecular mechanisms (Fig. 1).

NLRP3 inflammasome in the pathogenesis of hemorrhagic stroke. Whereas a priming signal such as the NF-κB pathway ensures the adequate gene expression of NLRP3, pro-IL 1β, and pro-IL 18 for efficient inflammasome formation, a second signal derived from damage-associated molecular pattern molecules, such as ATP and ROS, can trigger the activation of NLRP3 inflammasome to mediate neuroinflammation in the pathophysiological process of hemorrhagic stroke. The constitutive activation of the NLRP3 inflammasome promotes the processing and secretion of proinflammatory cytokines IL-1β and IL-18, which then leads to the recruitment of other immune cells to the affected brain tissue and results in the amplification of inflammatory responses following a hemorrhagic stroke
NLRP3 Inflammasome and Intracerebral Hemorrhage
An ICH is a common, albeit devastating, subtype of stroke (Qureshi et al. 2009). Secondary brain injury, which evolves over hours to days after an ICH, is a major determinant of the poor outcome in patients with an ICH (Keep et al. 2012). It is noteworthy that neuroinflammation plays a central role in the pathophysiology of an ICH (Wang 2010). Excessive inflammatory responses cause damage to the brain, which leads to cerebral edema and ongoing secondary brain injury after an ICH (Wang et al. 2013a). Therefore, elucidating the molecular basis of ICH-induced neuroinflammation is valuable for the identification of new therapeutic targets for this devastating disorder.
Recently, several lines of evidence have suggested that the constitutive activation of the NLRP3 inflammasome facilitates neuroinflammation via caspase-1 processing and IL-1β secretion following an ICH (Ma et al. 2014; Yang et al. 2015). Ma et al. demonstrated that NLRP3 inflammasome activation amplifies inflammatory responses, promotes neutrophil infiltration, exacerbates brain edema, and worsens neurological functions following an ICH in an autologous blood injection model in mice (Ma et al. 2014). They proposed that mitochondrial ROS may be a major trigger for the activation of NLRP3 inflammasome (Ma et al. 2014). Indeed, total ROS rather than mitochondrial ROS was measured in this study. Further studies are anticipated to explore the exact role and mechanism of mitochondrial ROS-induced NLRP3 inflammasome activation following an ICH. The results from another study have demonstrated that NLRP3 signaling is upregulated gradually from 1 to 5 days post-ICH in the perihematoma tissue (Yao et al. 2017). NLRP3 is required for the complement-induced neuroinflammation following an ICH, which plays an important role in ICH-induced neurological deficits (Yao et al. 2017). The exact component released from the ICH brain tissue that stimulates NLRP3 activation, however, is still unknown and must be further explored in future experiments. Using the TargetScan (version 4.2, www.targetscan.org) and Dual-Luciferase Reporter Assay system (promega, USA), Yang et al. found that microRNA-223 can directly regulate NLRP3 expression by targeting its 3′-untranslated region (3′-UTR) (Yang et al. 2015). MicroRNA-223 down-regulates the expression of NLRP3, inhibits inflammatory reaction, reduces brain edema, and improves neurological behaviors following experimental ICH in mice (Yang et al. 2015). Similarly, recombinant adenovirus encoding RNAi-mediated knockdown of an NLRP3 transcript inhibits an erythrocyte lysis-induced inflammatory response, decreases microglia-mediated neuronal injury, alleviates brain damage, and improves neurological outcomes following an ICH (Yuan et al. 2015). These findings suggest that the post-transcriptional regulation of NLRP3 expression by specific microRNA or RNAi may be a promising anti-inflammatory strategy in ICH. Additionally, it was found that the transmembrane P2X purinoceptor 7 (P2X7R) functions upstream of the NLRP3 activation and that it contributes to the production of NLRP3 inflammasome-dependent proinflammatory cytokines (e.g., IL-1β/IL-18) to drive brain inflammation and neuronal damage in a rat ICH model (Feng et al. 2015). An intracerebroventricular injection with a selective P2X7R inhibitor, blue brilliant G (BBG), down-regulates inflammatory responses by inhibiting the P2X7R/NLRP3 inflammasome axis following an ICH, which is beneficial to the reduction of cerebral edema and the improvement of functional outcomes after an ICH (Feng et al. 2015). These results indicate that the P2X7R-dependent synthesis of ONOO− may have an effect on triggering NLRP3 inflammasome activation (Feng et al. 2015). However, the exact mechanisms of ONOO− in modulating NLRP3 inflammasome activation have not been fully characterized. Ye et al. demonstrated that the ROS/TXNIP (thioredoxin binding protein) pathway contributes to thrombin-induced NLRP3 inflammasome activation and cell apoptosis in BV2 microglia with thrombin exposure (Ye et al. 2017b). However, the molecular basis underlying the activation of the NLRP3 inflammasome in an ICH-induced brain injury is not understood and warrants further investigation.
In conclusion, NLRP3 inflammasome-mediated inflammation contributes to secondary brain injury following an ICH, and the inhibition of NLRP3 inflammasome activation offers opportunities to limit inflammatory brain damage following an ICH.
NLRP3 Inflammasome and Subarachnoid Hemorrhage
An SAH is another life-threatening type of stroke with complex pathophysiological mechanisms (van Gijn et al. 2007). Once it occurs, a wide range of pathological processes (e.g., inflammation, apoptosis, oxidative stress) are initiated in the brain, which result in early brain injury (EBI) and delayed brain injury (DBI) following the event (Sehba et al. 2012; Macdonald 2014). More specifically, inflammation, which is a driving force behind the pathology of an SAH, contributes to both brain injury and cerebral vasospasm after an SAH (Sun et al. 2013; Vecchione et al. 2009).
As an important component of the innate immune system, the NLRP3 inflammasome has been proposed to play a vital role in the pathogenesis of SAH. For instance, Chen et al. demonstrated that the P2X7R/NLRP3 inflammasome axis is significantly activated 24 h after an SAH in an endovascular perforation rat model and is involved in the pathogenesis of an EBI after an SAH (Chen et al. 2013). The inhibition of P2X7R/NLRP3 inflammasome signaling via gene silencing of either cryopyrin or P2X7R suppresses caspase-1 activation and the subsequent production of mature IL-1β/IL-18 following an SAH, which in turn contributes to reduced brain edema and improved neurobehavioral functions following an SAH (Chen et al. 2013). Whether extracellular ATP engages P2X7R to trigger K+ efflux that leads to the assembly of the NLRP3 inflammasome after an SAH, however, has not been investigated in this study. Shao and colleagues found that a hydrogen-rich saline treatment attenuates the inflammation and cellular injury in an EBI by inhibiting NF-κB activation and NLRP3 inflammasome formation after an SAH (Shao et al. 2016). On the one side, a hydrogen-rich saline treatment inhibits the translocation of the NF-κB p65 subunit into the nucleus and decreases the gene expression of its downstream proinflammatory cytokines (e.g., IL-1β) and NLRP3 post-SAH. On the other side, a hydrogen-rich saline treatment reduces ROS generation, inhibits NLRP3 inflammasome activation, and decreases mature IL-1β production post-SAH. Li et al. reported that minocycline inhibits NLRP3 inflammasome activation and decreases neuroinflammation in an EBI after an SAH (Li et al. 2016). Minocycline treatment reduces cortical levels of ROS and alleviates NLRP3 inflammasome activation following an SAH (Li et al. 2016). Dong et al. found that melatonin treatment decreases ROS generation, inhibits NLRP3 inflammasome activation, and reduces the subsequent production of IL-1β and IL-18, thus providing neuroprotective effects against EBI following an SAH (Dong et al. 2016). Cao et al. reported that melatonin treatment promotes mitophagy, inhibits NLRP3 inflammasome activation, decreases proinflammatory cytokine levels, and protects against EBI after an SAH in an endovascular perforation rat model (Cao et al. 2017).
Together, these findings indicate that NLRP3 inflammasome-mediated neuroinflammation plays a crucial role in an EBI following an SAH. Hence, the blocking of NLRP3 inflammasome activation is beneficial for attenuating inflammatory responses following an SAH. However, no study has yet evaluated the role of the NLRP3 inflammasome in the development of cerebral vasospasm after an SAH, which is also an important cause of the poor outcomes for SAH patients. Accordingly, future studies that investigate the effect of the NLRP3 inflammasome on the pathogenesis of cerebral vasospasm following an SAH are anticipated.
NLRP3 Inflammasome and Hemorrhagic Transformation
A hemorrhagic transformation (HT), which is a bleeding into an area of an ischemic brain following a stroke, is a frequent complication of ischemic stroke and is exacerbated by thrombolytic therapy (Jickling et al. 2014). The experimental evidence suggests that multiple molecular mechanisms, such as increases in matrix metalloproteinases, oxidative stress, and inflammation, cause BBB disruption and result in an HT following an ischemic stroke (Bian et al. 2015; Imai et al. 2016; Kuroki et al. 2016). Recently, Guo et al. demonstrated that the NLRP3 inflammasome plays a key role in the pathologic process of an HT following MCAO in hyperglycemic rats (Guo et al. 2016). However, hyperbaric oxygen preconditioning prevents BBB disruption and attenuates hyperglycemia-enhanced HT by blocking the ROS/TXNIP/NLRP3 pathway (Guo et al. 2016). As these findings present pathophysiological significance and new therapeutic opportunities regarding the NLRP3 inflammasome in the pathogenesis of HT, they require further investigation.
Conclusion and Future Directions
The NLRP3 inflammasome is a member of the cytoplasmic multi-molecular complexes that controls the processing of caspase-1 and maturation of proinflammatory cytokines (e.g., IL-1β and IL-18). Diverse endogenous danger signals, such as ATP and ROS, can trigger the activation of the NLRP3 inflammasome to mediate neuroinflammation in the pathophysiological process of hemorrhagic stroke. The activation of the NLRP3 inflammasome generates high levels of inflammatory cytokines, which then leads to the recruitment of other immune cells to the affected tissue and the induction of inflammatory mechanisms to clear the DAMPs after hemorrhagic stroke. However, the overactivation of the NLRP3 inflammasome can result in sustained inflammation and brain damage following a hemorrhagic stroke. Thus, the investigation of the role of the NLRP3 inflammasome and the underlying mechanisms associated with hemorrhagic stroke will offer novel therapeutic strategies to treat this disease.
It is notable that a series of critical issues regarding the NLRP3 inflammasome in the pathogenesis of hemorrhagic stroke are unclarified and warrant examination. First, available evidence demonstrates that the microglia-expressed NLRP3 inflammasome is activated at high levels and plays an important role in the pathological processes of hemorrhagic stroke. However, whether neuron and astrocyte possess the NLRP3 inflammasome is still an unresolved question and requires further investigation. Furthermore, the mechanisms by which the NLRP3 inflammasome can sense different activators are not yet fully understood and require further elucidation. Furthermore, the activity and the role of the NLRP3 inflammasome during the late stage of hemorrhagic stroke are still unknown, but it is anticipated that they will be investigated. Answers to these questions are particularly important, as they will allow us to better manipulate this proinflammatory pathway and thereby alleviate the progression of hemorrhagic stroke.
References
Bian H, Hu Q, Liang X, Chen D, Li B, Tang J, Zhang JH (2015) Hyperbaric oxygen preconditioning attenuates hemorrhagic transformation through increasing PPARgamma in hyperglycemic MCAO rats. Exp Neurol 265:22–29. doi:10.1016/j.expneurol.2014.12.016
Bryant C, Fitzgerald KA (2009) Molecular mechanisms involved in inflammasome activation. Trends Cell Biol 19(9):455–464. doi:10.1016/j.tcb.2009.06.002
Brydges SD, Broderick L, McGeough MD, Pena CA, Mueller JL, Hoffman HM (2013) Divergence of IL-1, IL-18, and cell death in NLRP3 inflammasomopathies. J Clin Investig 123(11):4695–4705. doi:10.1172/JCI71543
Caner B, Hou J, Altay O, Fujii M, Zhang JH (2012) Transition of research focus from vasospasm to early brain injury after subarachnoid hemorrhage. J Neurochem 123(Suppl 2):12–21. doi:10.1111/j.1471-4159.2012.07939.x
Cao S, Shrestha S, Li J, Yu X, Chen J, Yan F, Ying G, Gu C, Wang L, Chen G (2017) Melatonin-mediated mitophagy protects against early brain injury after subarachnoid hemorrhage through inhibition of NLRP3 inflammasome activation. Sci Rep 7(1):2417. doi:10.1038/s41598-017-02679-z
Chen S, Ma Q, Krafft PR, Hu Q, Rolland W 2nd, Sherchan P, Zhang J, Tang J, Zhang JH (2013) P2X7R/cryopyrin inflammasome axis inhibition reduces neuroinflammation after SAH. Neurobiol Dis 58:296–307. doi:10.1016/j.nbd.2013.06.011
Chen L, Na R, Boldt E, Ran Q (2015) NLRP3 inflammasome activation by mitochondrial reactive oxygen species plays a key role in long-term cognitive impairment induced by paraquat exposure. Neurobiol Aging 36(9):2533–2543. doi:10.1016/j.neurobiolaging.2015.05.018
Dashdorj A, Jyothi KR, Lim S, Jo A, Nguyen MN, Ha J, Yoon KS, Kim HJ, Park JH, Murphy MP, Kim SS (2013) Mitochondria-targeted antioxidant MitoQ ameliorates experimental mouse colitis by suppressing NLRP3 inflammasome-mediated inflammatory cytokines. BMC Med 11:178. doi:10.1186/1741-7015-11-178
Dong Y, Fan C, Hu W, Jiang S, Ma Z, Yan X, Deng C, Di S, Xin Z, Wu G, Yang Y, Reiter RJ, Liang G (2016) Melatonin attenuated early brain injury induced by subarachnoid hemorrhage via regulating NLRP3 inflammasome and apoptosis signaling. J Pineal Res 60(3):253–262. doi:10.1111/jpi.12300
Fann DY, Lee SY, Manzanero S, Chunduri P, Sobey CG, Arumugam TV (2013) Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res Rev 12(4):941–966. doi:10.1016/j.arr.2013.09.004
Fann DY, Santro T, Manzanero S, Widiapradja A, Cheng YL, Lee SY, Chunduri P, Jo DG, Stranahan AM, Mattson MP, Arumugam TV (2014) Intermittent fasting attenuates inflammasome activity in ischemic stroke. Exp Neurol 257:114–119. doi:10.1016/j.expneurol.2014.04.017
Feng L, Chen Y, Ding R, Fu Z, Yang S, Deng X, Zeng J (2015) P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation 12:190. doi:10.1186/s12974-015-0409-2
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10(3):241–247. doi:10.1038/ni.1703
Guo ZN, Xu L, Hu Q, Matei N, Yang P, Tong LS, He Y, Guo Z, Tang J, Yang Y, Zhang JH (2016) Hyperbaric oxygen preconditioning attenuates hemorrhagic transformation through reactive oxygen species/thioredoxin-interacting protein/nod-like receptor protein 3 pathway in hyperglycemic middle cerebral artery occlusion rats. Crit Care Med 44(6):e403–411. doi:10.1097/CCM.0000000000001468
Hua Y, Keep RF, Hoff JT, Xi G (2007) Brain injury after intracerebral hemorrhage: the role of thrombin and iron. Stroke 38(2 Suppl):759–762. doi:10.1161/01.STR.0000247868.97078.10
Imai T, Takagi T, Kitashoji A, Yamauchi K, Shimazawa M, Hara H (2016) Nrf2 activator ameliorates hemorrhagic transformation in focal cerebral ischemia under warfarin anticoagulation. Neurobiol Dis 89:136–146. doi:10.1016/j.nbd.2016.02.001
Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, Morita R (2015) Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun 6:7360. doi:10.1038/ncomms8360
Jickling GC, Liu D, Stamova B, Ander BP, Zhan X, Lu A, Sharp FR (2014) Hemorrhagic transformation after ischemic stroke in animals and humans. J Cereb Blood Flow Metab 34(2):185–199. doi:10.1038/jcbfm.2013.203
Jo EK, Kim JK, Shin DM, Sasakawa C (2016) Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13(2):148–159. doi:10.1038/cmi.2015.95
Keep RF, Hua Y, Xi G (2012) Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol 11(8):720–731. doi:10.1016/S1474-4422(12)70104-7
Kleinig TJ, Vink R (2009) Suppression of inflammation in ischemic and hemorrhagic stroke: therapeutic options. Curr Opin Neurol 22(3):294–301
Kuroki T, Tanaka R, Shimada Y, Yamashiro K, Ueno Y, Shimura H, Urabe T, Hattori N (2016) Exendin-4 inhibits matrix metalloproteinase-9 activation and reduces infarct growth after focal cerebral ischemia in hyperglycemic mice. Stroke 47(5):1328–1335. doi:10.1161/STROKEAHA.116.012934
Lamkanfi M (2011) Emerging inflammasome effector mechanisms. Nat Rev Immunol 11(3):213–220. doi:10.1038/nri2936
Lamkanfi M, Dixit VM (2009) Inflammasomes: guardians of cytosolic sanctity. Immunol Rev 227(1):95–105. doi:10.1111/j.1600-065X.2008.00730.x
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13(6):397–411. doi:10.1038/nri3452
Lechtenberg BC, Mace PD, Riedl SJ (2014) Structural mechanisms in NLR inflammasome signaling. Curr Opin Struct Biol 29:17–25. doi:10.1016/j.sbi.2014.08.011
Leemans JC, Cassel SL, Sutterwala FS (2011) Sensing damage by the NLRP3 inflammasome. Immunol Rev 243(1):152–162. doi:10.1111/j.1600-065X.2011.01043.x
Li J, Yan F, Chen G (2014) Reactive oxygen species and NLRP3 inflammasome activation. Ann Neurol 75(6):972. doi:10.1002/ana.24173
Li J, Chen J, Mo H, Chen J, Qian C, Yan F, Gu C, Hu Q, Wang L, Chen G (2016) Minocycline protects against NLRP3 inflammasome-induced inflammation and P53-associated apoptosis in early brain injury after subarachnoid hemorrhage. Mol Neurobiol 53(4):2668–2678. doi:10.1007/s12035-015-9318-8
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH (2014) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156(6):1193–1206. doi:10.1016/j.cell.2014.02.008
Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J (2014) NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol 75(2):209–219. doi:10.1002/ana.24070
Macdonald RL (2014) Delayed neurological deterioration after subarachnoid haemorrhage. Nat Rev Neurol 10(1):44–58. doi:10.1038/nrneurol.2013.246
Mariathasan S, Monack DM (2007) Inflammasome adaptors and sensors: intracellular regulators of infection and inflammation. Nat Rev Immunol 7(1):31–40. doi:10.1038/nri1997
Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G (2013) K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38(6):1142–1153. doi:10.1016/j.immuni.2013.05.016
Proell M, Gerlic M, Mace PD, Reed JC, Riedl SJ (2013) The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem J 449(3):613–621. doi:10.1042/BJ20121198
Qureshi AI, Wilson DA, Hanley DF, Traystman RJ (1999) No evidence for an ischemic penumbra in massive experimental intracerebral hemorrhage. Neurology 52(2):266–272
Qureshi AI, Mendelow AD, Hanley DF (2009) Intracerebral haemorrhage. Lancet 373(9675):1632–1644. doi:10.1016/S0140-6736(09)60371-8
Rinkel GJ, Algra A (2011) Long-term outcomes of patients with aneurysmal subarachnoid haemorrhage. Lancet Neurol 10(4):349–356. doi:10.1016/S1474-4422(11)70017-5
Schallner N, Pandit R, LeBlanc R 3rd, Thomas AJ, Ogilvy CS, Zuckerbraun BS, Gallo D, Otterbein LE, Hanafy KA (2015) Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Invest 125(7):2609–2625. doi:10.1172/JCI78443
Segovia J, Sabbah A, Mgbemena V, Tsai SY, Chang TH, Berton MT, Morris IR, Allen IC, Ting JP, Bose S (2012) TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 7(1):e29695. doi:10.1371/journal.pone.0029695
Sehba FA, Hou J, Pluta RM, Zhang JH (2012) The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol 97(1):14–37. doi:10.1016/j.pneurobio.2012.02.003
Shao A, Wu H, Hong Y, Tu S, Sun X, Wu Q, Zhao Q, Zhang J, Sheng J (2016) Hydrogen-rich saline attenuated subarachnoid hemorrhage-induced early brain injury in rats by suppressing inflammatory response: possible involvement of NF-kappaB pathway and NLRP3 inflammasome. Mol Neurobiol 53(5):3462–3476. doi:10.1007/s12035-015-9242-y
Sun X, Ji C, Hu T, Wang Z, Chen G (2013) Tamoxifen as an effective neuroprotectant against early brain injury and learning deficits induced by subarachnoid hemorrhage: possible involvement of inflammatory signaling. J Neuroinflammation 10:157. doi:10.1186/1742-2094-10-157
Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10(3):210–215. doi:10.1038/nri2725
van Gijn J, Kerr RS, Rinkel GJ (2007) Subarachnoid haemorrhage. Lancet 369(9558):306–318. doi:10.1016/S0140-6736(07)60153-6
Vecchione C, Frati A, Di Pardo A, Cifelli G, Carnevale D, Gentile MT, Carangi R, Landolfi A, Carullo P, Bettarini U, Antenucci G, Mascio G, Busceti CL, Notte A, Maffei A, Cantore GP, Lembo G (2009) Tumor necrosis factor-alpha mediates hemolysis-induced vasoconstriction and the cerebral vasospasm evoked by subarachnoid hemorrhage. Hypertension 54(1):150–156. doi:10.1161/HYPERTENSIONAHA.108.128124
Wang J (2010) Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol 92(4):463–477. doi:10.1016/j.pneurobio.2010.08.001
Wang J, Dore S (2007) Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab 27(5):894–908. doi:10.1038/sj.jcbfm.9600403
Wang YC, Wang PF, Fang H, Chen J, Xiong XY, Yang QW (2013a) Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke 44(9):2545–2552. doi:10.1161/STROKEAHA.113.001038
Wang Z, Wu L, You W, Ji C, Chen G (2013b) Melatonin alleviates secondary brain damage and neurobehavioral dysfunction after experimental subarachnoid hemorrhage: possible involvement of TLR4-mediated inflammatory pathway. J Pineal Res 55(4):399–408. doi:10.1111/jpi.12087
Wang YC, Zhou Y, Fang H, Lin S, Wang PF, Xiong RP, Chen J, Xiong XY, Lv FL, Liang QL, Yang QW (2014) Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol 75(6):876–889. doi:10.1002/ana.24159
Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, Hoffman HM, Feldstein AE (2014) NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 59(3):898–910. doi:10.1002/hep.26592
Xi G, Keep RF, Hoff JT (1998) Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J Neurosurg 89(6):991–996. doi:10.3171/jns.1998.89.6.0991
Xi G, Keep RF, Hoff JT (2006) Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol 5(1):53–63. doi:10.1016/S1474-4422(05)70283-0
Xi G, Strahle J, Hua Y, Keep RF (2014) Progress in translational research on intracerebral hemorrhage: is there an end in sight? Prog Neurobiol 115:45–63. doi:10.1016/j.pneurobio.2013.09.007
Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, Shentu TP, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JH, Johnson DA, Shyy JY (2013) Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 128(6):632–642. doi:10.1161/CIRCULATIONAHA.113.002714
Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, Guarda G, Tian Z, Tschopp J, Zhou R (2013) Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 38(6):1154–1163. doi:10.1016/j.immuni.2013.05.015
Yang F, Wang Z, Wei X, Han H, Meng X, Zhang Y, Shi W, Li F, Xin T, Pang Q, Yi F (2014) NLRP3 deficiency ameliorates neurovascular damage in experimental ischemic stroke. J Cereb Blood Flow Metab 34(4):660–667. doi:10.1038/jcbfm.2013.242
Yang Z, Zhong L, Xian R, Yuan B (2015) MicroRNA-223 regulates inflammation and brain injury via feedback to NLRP3 inflammasome after intracerebral hemorrhage. Mol Immunol 65(2):267–276. doi:10.1016/j.molimm.2014.12.018
Yao ST, Cao F, Chen JL, Chen W, Fan RM, Li G, Zeng YC, Jiao S, Xia XP, Han C, Ran QS (2017) NLRP3 is required for complement-mediated caspase-1 and IL-1beta activation in ICH. J Mol Neurosci 61(3):385–395. doi:10.1007/s12031-016-0874-9
Ye X, Shen T, Hu J, Zhang L, Zhang Y, Bao L, Cui C, Jin G, Zan K, Zhang Z, Yang X, Shi H, Zu J, Yu M, Song C, Wang Y, Qi S, Cui G (2017a) Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp Neurol 292:46–55. doi:10.1016/j.expneurol.2017.03.002
Ye X, Zuo D, Yu L, Zhang L, Tang J, Cui C, Bao L, Zan K, Zhang Z, Yang X, Chen H, Tang H, Zu J, Shi H, Cui G (2017b) ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem Biophys Res Commun 485(2):499–505. doi:10.1016/j.bbrc.2017.02.019
Yuan B, Shen H, Lin L, Su T, Zhong S, Yang Z (2015) Recombinant adenovirus encoding NLRP3 RNAi attenuate inflammation and brain injury after intracerebral hemorrhage. J Neuroimmunol 287:71–75. doi:10.1016/j.jneuroim.2015.08.002
Zaki MH, Lamkanfi M, Kanneganti TD (2011) The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol 32(4):171–179. doi:10.1016/j.it.2011.02.002
Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, Grotta JC, Aronowski J (2007) Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol 61(4):352–362. doi:10.1002/ana.21097
Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW (2014) Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol 115:25–44. doi:10.1016/j.pneurobio.2013.11.003
Acknowledgements
This work was supported by the Zhejiang Provincial Administration of Traditional Chinese Medicine (2016ZQ004).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Yang, SJ., Shao, GF., Chen, JL. et al. The NLRP3 Inflammasome: An Important Driver of Neuroinflammation in Hemorrhagic Stroke. Cell Mol Neurobiol 38, 595–603 (2018). https://doi.org/10.1007/s10571-017-0526-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-017-0526-9