
Abstract
Candesartan has been reported to have a protective effect on cerebral ischemia in vivo and in human ischemic stroke. We studied the direct effects of candesartan on blood–brain barrier (BBB) function with our in vitro monolayer model generated using rat brain capillary endothelial cells (RBECs). The in vitro BBB model was subjected to normoxia or 6-h oxygen glucose deprivation (OGD)/24-h reoxygenation, with or without candesartan. 6-h OGD/24-h reoxygenation decreased transendothelial electrical resistance and increased the endothelial permeability for sodium fluorescein in RBEC monolayers. Candesartan (10 nM) improved RBEC barrier dysfunction induced by 6-h OGD/24-h reoxygenation. Immunostaining and immunoblotting analysis indicated that the effect of candesartan on barrier function under 6-h OGD/24-h reoxygenation was not related to the expression levels of tight junction proteins. However, candesartan affected RBEC morphological changes induced by 6-h OGD/24-h reoxygenation. We analyzed oxidative stress and cell viability using chemical reagents. Candesartan improved cell viability following 6-h OGD/24-h reoxygenation, whereas candesartan had no effect on oxidative stress. These results show that candesartan directly improves cell function and viability of brain capillary endothelial cells under OGD/reoxygenation, suggesting that the protective effects of candesartan on ischemic stroke are related to protection of the BBB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The renin–angiotensin system (RAS) plays an important role in the regulation of fluid volume and blood pressure, and is closely connected with cardiovascular diseases such as hypertension, heart failure, and stroke (Zaman et al. 2002; Schmieder et al. 2007). The angiotensin II (Ang II) has various influences on vascular systems via its specific receptors (AT1 and AT2) under pathological and physiological conditions. AT1 receptor blockers (ARBs) specifically inhibit the diverse physiological effects mediated by the AT1 receptor. Thus, AT1 receptor blockade is highly relevant to the treatment of cardiovascular disease. ARBs not only have strong blood pressure-lowering effects in hypertensive patients, but also induce regression of left ventricular hypertrophy and decrease cardiovascular morbidity and mortality in patients with heart failure (Cohn et al. 2001; Lindholm et al. 2002; Pfeffer et al. 2003).
Cerebral ischemia, a leading cause of death and disability, induces cerebral vascular and neuronal damage. Several recent clinical trials have shown the efficacy of ARBs in primary and secondary prevention of stroke in patients with hypertension and/or elevated cardiovascular risk (Dahlöf et al. 2002; Schrader et al. 2003; Ogihara et al. 2008). In addition, treatment of patients with the ARB candesartan during the acute phase of cerebral ischemia produced beneficial effects on cerebrovascular events that were independent of alterations in blood pressure (Schrader et al. 2003). As the RAS components exist in vascular systems as well as in the central nervous system, candesartan appears to affect both vascular and neural systems. Indeed, several reports have indicated that candesartan improves neuronal damage and cognitive impairment caused by cerebral ischemia (Lu et al. 2005; Skoog et al. 2005). In contrast, the consequences of AT1 receptor blockade on the cerebral vascular system are controversial. Although AT1 receptor blockade decreased vascular hyperpermeability induced by experimental stroke, (Kozak et al. 2009, Panahpour et al. 2014), Wosik et al. (2007) reported that AT1 stimulation by Ang II strengthens blood–brain barrier (BBB) function under normal conditions. Thus, we aimed to investigate the effects of AT1 receptor blockade on BBB barrier function under normal and hypoxic conditions.
Brain capillary endothelial cells form the anatomical basis of the BBB and have a highly specialized system for regulating the passage of substances from the blood into the brain parenchyma (Engelhardt 2003; Abbott 2005). As the brain capillary endothelial cells are tightly sealed with adhesion molecules present in tight junctions and adherens junctions, vascular damage induced by cerebral stroke results in the development of brain edema. In addition, BBB dysfunction allows inflammatory cell penetration into the brain parenchyma, and these insults may be correlated with the acceleration of neurological damage (Edvinsson and Povlsen 2011; Dirnagl 2012). Thus, protecting BBB integrity during cerebral stroke has a strong influence on preventing cerebral damage. However, whether AT1 blockade affects BBB barrier function in vitro is not well understood.
In this study, we hypothesized that candesartan has the potential to protect the BBB during cerebral stroke. We have investigated the effects of candesartan on rat brain capillary endothelial cell dysfunction induced by an oxygen and glucose deprivation model in vitro, and explored the mechanism involved.
Materials and Methods
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise indicated. Wistar rats were obtained from Japan SLC, Inc. (Japan). All animals were treated in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) and approved by the Nagasaki University Animal Care Committee. Candesartan (Blopress®) was provided by Takeda Pharmaceutical Co., Ltd., Japan.
Cell Cultures
Rat Brain Capillary Endothelial Cells (RBECs)
Primary cultures of rat brain capillary endothelial cells (RBECs) were prepared from 3-week-old rats, as previously described (Nakagawa et al. 2007, 2009). In brief, meninges were carefully removed from forebrains and gray matter was minced. The homogenate was digested with collagenase type 2 (1 mg/ml, Worthington Biochemical Corp., USA) and DNase (15 μg/ml) in Dulbecco’s modified Eagle’s medium (DMEM) (WAKO Pure Chemical Ltd., Japan) for 1.5 h at 37 °C. The cell pellet was separated by centrifugation in 20 % bovine serum albumin (BSA)-DMEM (1,000×g, 20 min). The microvessels obtained in the pellet were further digested with collagenase-dispase (1 mg/ml; Roche Applied Sciences, Rotkreuz, Switzerland) and DNase (6.7 μg/ml) in DMEM for 30 min at 37 °C. Microvessels were separated on a 33 % continuous Percoll (GE Healthcare, Uppsala, Sweden) gradient. Microvessels were then seeded on plastic dishes coated with collagen type IV and fibronectin (both 0.1 mg/ml). RBEC cultures were maintained in DMEM/F12 supplemented with 10 % fetal bovine plasma derived serum (PDS) (Animal Technologies, Inc., Tyler, TX, USA), basic fibroblast growth factor (bFGF, 1.5 ng/ml; Roche Applied Sciences), heparin (100 μg/ml), insulin (5 μg/ml), transferrin (5 μg/ml), sodium selenite (5 ng/ml) (insulin–transferrin–sodium selenite media supplement), gentamycin (50 μg/ml), and puromycin (4 μg/ml) (RBEC I medium) at 37 °C for 2 days. The culture medium was then replaced with fresh culture medium containing all the components of RBEC I medium except for puromycin. The cultures reached 80 % confluence (day 4) and purified endothelial cells were collected by brief treatment with trypsin (0.05 % w/v)-EDTA (0.02 % w/v) solution, and were used to construct the in vitro BBB model.
Rat Cerebral Astrocytes
Rat cerebral astrocytes were obtained from neonatal Wistar rats. Meninges were removed and cortical pieces mechanically dissociated in DMEM. Dissociated cells were passed through a 70-μm nylon mesh (BD Falcon, MA, USA) and seeded into cell culture flasks. Astrocytes cultures were maintained in culture medium (DMEM supplemented with 10 % fetal bovine serum). In order to obtain type 1 astrocytes as pure as possible, flasks with confluent cultures were shaken strongly by hand to remove microglia. This procedure was performed three times during a 2-week culture period. The purity of astrocytes was checked by immunostaining for glial fibrillary acidic protein (GFAP), and the cells were used at passage 2–3.
In Vitro BBB Model
An in vitro BBB model was constructed with Transwell® (Corning Incorporated Life Sciences, NY, USA) inserts in 12-well culture plates. Endothelial cells (1.5 × 105 cells/cm2) were seeded on the upper side of the collagen- and fibronectin-coated polyester membranes (0.4-μm pore size) of the Transwell® inserts. The in vitro BBB model was maintained in RBEC medium II for 1 day, with the medium subsequently changed to RBEC medium II containing 500 nM hydrocortisone. To construct a co-cultured BBB model, astrocytes (1.5 × 105 cells/well) were seeded on the bottom of the collagen coated of 12-well culture plate (Costar, Corning, NY, USA). The cells were allowed to adhere firmly overnight. Thereafter, RBECs (1.5 × 105 cells/cm2) were seeded in the inserts, as described above.
Candesartan Treatment
The candesartan stock solution (10 mM) was stored at −20 °C, and diluted in medium for the experiments. To compare the effects of candesartan on the RBEC monolayer model versus the co-culture model, candesartan was added to both the luminal and abluminal sides of the BBB models. In all other experiments, candesartan was added to the luminal surface. Based on previous reports, we used a dose of 10 nM of candesartan (Kataoka et al. 2010; Soliman et al. 2014).
Normoxia Conditions
Cells were transferred into serum-free RBEC medium II, with or without candesartan. Following 6 h of candesartan treatment, the medium was exchanged to RBEC medium II with or without candesartan and the cells were incubated for a further 24 h.
Oxygen and Glucose Deprivation (OGD) and Reoxygenation Conditions
Consistent with our previous reports (Hiu et al. 2008, Horai et al. 2013), oxygen and glucose deprivation (OGD)/reoxygenation conditions consisted of 6-h OGD and 24-h reoxygenation. For 6-h OGD treatments, serum-free and glucose-free DMEM (Gibco, Carlsbad, CA, USA) with or without candesartan were added to the BBB model. Oxygen deprivation conditions were generated using ANAEROPACK® (Mitsubishi Gas Chemical Co., Inc., Tokyo, Japan). For 6-h normoxic groups, serum-free and 17.5 mM glucose DMEM with or without candesartan were added to the BBB model. Reoxygenation was initiated by adding fresh RBEC II medium with or without candesartan after 6-h OGD or 6-h normoxia.
Evaluation of Barrier Function in RBEC
Transendothelial Electrical Resistance (TEER)
Transendothelial electrical resistance (TEER) was measured using an EVOM resistance meter (World Precision Instruments, Sarasota, FL, USA). The resistance measurements of blank filters were subtracted from those of filters with cells. The values are shown as Ω cm2 based on culture inserts.
Paracellular Permeability of Sodium Fluorescein
Endothelial barrier function was evaluated by measuring the permeability of sodium fluorescein (Na–F, MW 376). To initiate the permeability studies, the medium in the luminal chamber of the insert (0.5 ml) was removed and replaced with assay buffer (136 mM NaCl, 0.9 mM CaCl2, 0.5 mM MgCl2, 2.7 mM KCl, 1.5 mM KH2PO4, 10 mM NaH2PO4, 25 mM glucose, and 10 mM Hepes, pH 7.4) containing 10 μg/ml Na–F. Samples were removed from the abluminal chamber at 30 min. The concentration of Na–F was determined using a fluorescence multi-well plate reader [E x (λ) 485 nm; E m (λ) 535 nm]. The permeability coefficient was calculated as previously described (Youdim et al. 2003).
Immunostaining
For immunofluorescence, cells were fixed in 4 % paraformaldehyde for 20 min and permeabilized with 0.1 % Triton X-100 for 10 min. After washing with phosphate buffered saline (PBS) and blocking with 3 % bovine serum albumin (BSA) in PBS for 30 min., the samples were stained with primary antibodies for claudin-5 and occludin (Invitrogen Corporation, Carlsbad, CA, USA). All primary antibodies were used at a dilution of 1:200. The secondary antibody, Alexa Flour 488 conjugated donkey anti-mouse immunoglobulin (Invitrogen Corporation), was used at a dilution of 1:1,000. The samples were mounted in Gel Mount (Biomeda, Foster City, CA, USA) and staining was assessed using a Zeiss LSM 5 Pascal Confocal laser-scanning microscope (Carl Zeiss AG, Oberkochen, Germany).
Immunoblotting
Cells were lysed using the CelLytic™ M cell lysis reagent supplemented with proteinase inhibitor cocktail (Sigma) and centrifuged at 15,000×g for 15 min. The supernatants were collected and protein levels were determined using a BCA protein assay kit (Pierce, Rockford, IL, USA). Samples were separated on 4–15 % Tris–HCl Ready SDS–polyacrylamide gel (Bio-Rad Laboratories, Hercules, CA, USA), transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories), and incubated with primary antibodies. Antibodies were diluted 1:2,500 (occludin), 1:5,000 (claudin-5), and 1:10,000 (β-actin) in blocking solution (1 % Perfect-Block, MoBiTec, Germany). Peroxidase-conjugated anti-mouse immunoglobulin (GE Healthcare) was used as a secondary antibody. To reveal immunoreactive bands, blots were incubated in SuperSignal West Femto Maximum Sensitivity Substrate SECL in accordance with the manufacturer’s instructions (Pierce Biotechnology, Rockford, IL, USA) and were detected using a FluorChem SP Imaging System (Alpha Innotech Corp., San Leandro, CA, USA).
Nitrate/Nitrite Levels in the RBEC Culture Medium
To measure the amounts of nitrate (NO2)/nitrite (NO3), culture medium was collected after 6-h OGD or 6-h OGD/24-h reoxygenation. The amounts of NO2/NO3 were measured using a high-performance liquid chromatography (HPLC) system (NOx analyzer ENO-10, EICOM, Kyoto, Japan) according to Yamada and Nabeshima (1997). In brief, NO2 and NO3 in culture medium were separated using a reverse-phase separation column (NO-PAK, EICOM) and NO3 was reduced to NO2 in a reduction column (NO-RED, EICOM). NO2 was mixed with Griess reagent to form a purple azo dye in a reaction coil, and the resulting dye was measured using a flow-through spectrophotometer.
Intracellular Levels of Reactive Oxygen Species (ROS)
Intracellular ROS were measured with a CM-H2DCFDA (Invitrogen). CM-H2DCFDA is a cell-permeable fluorescent dye which generates fluorescence upon reaction with ROS. RBECs were seeded at a density of 5,000 cells per well into 96-well plates. After confluency, RBECs were exposed to 6-h OGD or 6-h OGD/24-h reoxygenation. Subsequently, CM-H2DCFDA (5 μM) in assay buffer was added and further incubated for 30 min. The fluorescence intensity of the probe was determined using a fluorescence multi-well plate reader (E x (λ) 485 nm; E m (λ) 535 nm). The values of ROS produced are presented as a percent of control.
Cell Viability
Rat brain capillary endothelial cells (RBECs) were seeded at a density of 5,000 cells per well into 96-well plates in culture medium. After exposure to 6-h OGD or 6-h OGD/24-h reoxygenation, the number of viable cells was determined using a Cell Counting Kit 8 (Dojindo Co., Kumamoto, Japan) according to the manufacturer’s instructions. The assay reagent is a tetrazolium compound (WST-8) that is reduced by live cells into a colored formazan product measured at 450 nm.
Statistical Analysis
All data presented are mean ± standard error of the mean (SEM). The values were compared using an analysis of variance followed by the Tukey–Kramer post hoc test. Changes were considered statistically significant at P < 0.05.
Results
To investigate whether candesartan could affect the integrity of endothelial monolayers under normal culture conditions, two BBB models, RBEC monolayer model and RBEC co-cultured with astrocytes, were treated with candesartan (10 nM) and barrier function was measured using transendothelial electrical resistance (TEER). In agreement with previous reports, barrier function in cultured brain endothelial cells was enhanced by co-culture with astrocytes (control value at 72 h: 150.8 ± 6.5 vs. 264.0 ± 20.0 Ω cm2) (Fig. 1a, b). Although candesartan did not affect TEER in the RBEC monolayer model, candesartan enhanced TEER in the RBEC-astrocytes co-culture model (Fig. 1a, b).

Transendothelial electrical resistance (TEER) in two in vitro blood–brain barrier models. Rat brain capillary endothelial cells (RBECs) were seeded on transwell inserts 1 day before candesartan was added to both the luminal and abluminal sides of the inserts. a RBEC monolayers was exposed to 10 nM candesartan under normal cell culture conditions. Candesartan did not affect TEER in the monolayer model. b RBEC-astrocyte co-cultures were exposed to 10 nM candesartan under normal cell culture conditions. Candesartan increased TEER in RBECs co-cultured with astrocytes. All data are presented as mean ± SEM (n = 8). *P < 0.05, significant difference from control
Although several reports indicate that candesartan is protective toward ischemic damage, potential direct effects of candesartan on endothelial cells remain to be elucidated. To focus on the effects of candesartan on brain capillary endothelial cells, we used the RBEC monolayer model to eliminate the influence of astrocytes.
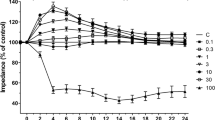
In order to analyze the potential beneficial effects of candesartan on ischemic endothelial damage, RBECs were subjected to 6-h OGD/24-h reoxygenation. Endothelial barrier function was estimated by measuring TEER and paracellular permeability of Na–F (Fig. 2). TEER was significantly decreased following 6-h OGD/24-h reoxygenation compared to normoxia (86.8 ± 2.2 vs. 100 ± 2.1 %; P < 0.05). The 6-h OGD/24-h reoxygenation-induced decrease in TEER was significantly suppressed by candesartan treatment (86.8 ± 2.2 vs. 97.9 ± 2.6 %; P < 0.05) (Fig. 2a). Disruption of barrier integrity was also detected by determining Na–F permeability in RBECs following 6-h OGD/24-h reoxygenation compared with normoxia (4.81 ± 0.4 vs. 3.17 ± 0.2 × 10−6 cm/s, P < 0.01). Treatment of the cells subjected to 6-h OGD/24-h reoxygenation with 10 nM candesartan produced Na–F permeability similar to the normoxia control value (Fig. 2b).

Effects of candesartan on barrier function in rat brain capillary endothelial cell (RBEC) monolayers under normoxia and 6-h oxygen glucose deprivation (OGD)/24-h reoxygenation. Barrier function was evaluated by transendothelial electrical resistance (TEER) (a) and sodium fluorescein (Na–F) permeability (b). a TEER was significantly decreased in RBEC with 6-h OGD/24-h reoxygenation. Candesartan attenuated the 6-h OGD/24-h reoxygenation-induced decrease in TEER. Data indicate the rate of change in TEER before and after treatment in comparison with normoxia control (100 % = 101.8 ± 3.2 Ω × cm2, n = 9). All data are presented as mean ± SEM (n = 7–11). *P < 0.05, significant difference from normoxia control. # P < 0.05, significant difference from 6-h OGD/24-h reoxygenation control. b Na–F permeability was significantly increased in RBEC with 6-h OGD/24-h reoxygenation. Candesartan attenuated the 6-h OGD/24-h reoxygenation-induced increase in Na–F permeability. All data are presented as mean ± SEM (n = 9–15). **P < 0.01, significant difference from normoxia control. ## P < 0.01, significant difference from 6-h OGD/24-h reoxygenation control
To examine whether candesartan affects the expression of tight junction proteins, which is a factor in regulating barrier function, we performed immunostaining and immunoblotting. There were no notable changes in the appearance of claudin-5 and occludin following 6-h OGD/24-h reoxygenation (Fig. 3b, e) compared with the normoxia control (Fig. 3a, d). In agreement with immunostaining observations, claudin-5 and occludin protein levels, determined by immunoblotting, were not altered by 6-h OGD/24-h reoxygenation. Although 6-h OGD/24-h reoxygenation did not affect the expression of tight junction proteins, alterations in cellular morphology were detected following 6-h OGD/24-h reoxygenation. 6-h OGD/24-h reoxygenation induced a cobble stone morphology that was not observed under normoxia. Candesartan treatment suppressed the effects of 6-h OGD/24-h reoxygenation on endothelial cellular morphology (Fig. 3c, f).

Expression of tight junction proteins in rat brain capillary endothelial cell (RBEC) monolayers. The expression of claudin-5 (a–c) and occludin (d–f) was determined by immunostaining of RBEC monolayers. Although there was no obvious change in the expression of claudin-5 and occludin in 6-h OGD/24-h reoxygenation (b, e) compared with cells under normoxia (a, d), 6-h OGD/24-h reoxygenation changed the cellular morphology to a cobble stone appearance (b, e). Candesartan (10 nM) suppressed the 6-h OGD/24-h reoxygenation-induced changes in cellular morphology (c, f). Bar 20 μm. The levels of claudin-5 (g) and occludin (h) protein expression were determined by immunoblotting. No significant changes were detected in each group. The blots are representative images from separate experiments. Data are presented as mean ± SEM (n = 6)
As oxidative stress is a factor that induces cellular damage under OGD/reoxygenation, we analyzed the effects of candesartan on the production of nitrate/nitrite and ROS. There was no significant difference in nitrate/nitrite levels as a result of 6-h OGD (Fig. 4a). Nitrate/nitrite level in the RBEC culture medium was significantly higher following 6-h OGD/24-h reoxygenation compared to normoxia (0.46 ± 0.01 vs. 0.41 ± 0.01 × 10−5 M; P < 0.05) (Fig. 4b). Although candesartan tended to suppress the elevated nitrate/nitrite levels induced by 6-h OGD/24-h reoxygenation, significant differences were not detected. ROS production was significantly higher after 6-h OGD than normoxia (233.9 ± 15.3 vs. 100 ± 13.4 %; P < 0.01) (Fig. 4c). However, increased ROS production was not detected after an additional 24-h of reoxygenation (Fig. 4d). Candesartan did not affect the ROS production induced by 6-h OGD.

Effects of candesartan on the production of nitrate/nitrite and ROS. Following 6-h OGD (a, c) or 6-h OGD/24-h reoxygenation (b, d), production of nitrate/nitrite and ROS was determined using the Griess reagent and a fluorogenic dye, respectively. Although the levels of nitrate/nitrite were not altered by 6-h OGD alone (a), 6-h OGD/24-h reoxygenation significantly increased the production of nitrate/nitrite into the culture medium (b). Candesartan did not affect the production of nitrate/nitrite. All data are presented as mean ± SEM (n = 4). *P < 0.05, significant difference from normoxia control. c The levels of ROS in rat brain capillary endothelial cell (RBEC) monolayers were increased following 6-h OGD. d ROS levels in RBECs 24 h after reoxygenation were equivalent in all groups. Candesartan did not affect ROS production. ROS production is expressed as a percentage of the normoxia control values. All data are presented as mean ± SEM (n = 4–5). **P < 0.01, significant difference from normoxia control
Lastly, we examined the effects of candesartan on cell viability following 6-h OGD/24-h reoxygenation. There was no significant difference in endothelial cell viability among the groups following 6-h OGD (Fig. 5a). In contrast, 6-h OGD/24-h reoxygenation decreased cell viability. The decreased cell viability induced by 6-h OGD/24-h reoxygenation was improved by candesartan treatment (Fig. 5b).

Effects of candesartan on cell viability in rat brain capillary endothelial cells (RBECs). Following a 6-h OGD (a) or 6-h OGD/24-h reoxygenation (b), cell viability was determined using a cell counting Kit-8. There was no significant difference in endothelial cell viability after 6-h OGD (a). Candesartan inhibited the 6-h OGD/24-h reoxygenation-induced decreases in cell viability. Cell viability is expressed as a percentage of the normoxia control values. All data are presented as mean ± SEM (n = 5–14). **P < 0.01, significant difference from normoxia control. ## P < 0.01, significant difference from 6-h OGD/24-h reoxygenation control
Discussion
BBB dysfunction resulting from cerebral stroke causes brain edema formation, and increases the penetration of neurotoxic factors into the brain parenchyma. Thus, improving BBB function during cerebral stroke has a significant potential to prevent the progression of neuronal damage. In the present study, we found that candesartan prevents BBB dysfunctions in an in vitro ischemia model involving 6-h OGD/24-h reoxygenation. Our present observations support previous studies showing that candesartan reduces brain damage induced by cerebral ischemia, and attenuates learning dysfunction in a rat ischemia model (Lu et al. 2005; Faure et al. 2008; Kozak et al. 2009).
The renin-angiotensin system (RAS) plays an important role in regulating the cardiovascular and central nervous systems (Saavedra 2012). There are controversial reports regarding the effects of angiotensin II (Ang II), mediated via its receptors (AT1 and AT2), on the barrier function of brain endothelial cells under physiological conditions. Several reports indicate that activation of AT1 receptors increases endothelial permeability (Fleegal-DeMotta et al. 2008; Zhang et al. 2010; Pelisch et al. 2011). However, Wosik et al. (2007) indicated that Ang II, secreted by astrocytes, improved BBB integrity. Therefore, we examined the effects of candesartan on BBB barrier function under normal conditions. Our finding showed that candesartan did not strengthen the barrier properties of RBEC monolayers under normal culture conditions up to 72 h. In contrast, candesartan increased TEER in RBEC co-cultured with astrocytes. It seems that astrocyte-derived Ang II decreases BBB barrier function, and candesartan inhibited this effect in our rodent BBB model. Wosik et al. (2007) used a large molecule (FITC-labeled bovine serum albumin) as a permeability tracer, together with human brain endothelial cells derived from patients with intractable epilepsy as a BBB model. In contrast, we used a small molecule (sodium fluorescein) and endothelial cells derived from normal rats. Considering that we could not detect BSA leakage under basal conditions in our rodent BBB model (data not shown), it appears that the human BBB model has a relatively leaky barrier. Therefore, the effects of AT1 stimulation of endothelial cells may have different consequences on the barrier function depending on factors such as cell type and cell viability. More studies to characterize AT1-induced signaling are required to fully understand these observations.
Next, to investigate the effect of AT1 blockade under pathological conditions, we examined the effects of candesartan on RBEC barrier properties under OGD/reoxygenation. In this study, we used a RBEC monolayer model to focus on the effects of candesartan on endothelial cells alone. We established a pathophysiological in vitro BBB model by exposing endothelial cells to normoxia or 6-h OGD/24-h reoxygenation. The results showed that OGD/reoxygenation increased the permeability of Na–F and decreased TEER. We demonstrated that in this in vitro monolayer model, candesartan (10 nM) inhibited the OGD/reoxygenation-induced changes in TEER and NaF permeability, which suggests that candesartan may protect against deficits in paracellular transport following OGD/reoxygenation.
As TEER and endothelial cell permeability reflect tight junction (TJ) function (Deli et al. 2005), we examined the effect of candesartan on TJ protein levels using immunostaining and Western blotting. However, there were no significant differences in the expression of caludin-5 and occludin after candesartan treatment. Thus, the effect of candesartan on BBB permeability is not associated with changes in the expression of tight junction proteins. Although reductions in expression of tight junction proteins lead to increased permeability, the expression pattern of tight junction proteins along cell–cell borders and the phosphorylation of tight junction proteins are important factors in modulating endothelial permeability (Ishizaki et al. 2003; Yamamoto et al. 2008). Since it remains possible that candesartan may influence claudin-5 and occludin without altering protein levels, further studies are needed. Alternatively, as candesartan suppressed OGD/reoxygenation-induced alterations in cellular morphology, it is possible that candesartan may affect the cytoskeletal protein actin, because rearrangement of cytoskeletal actin is known to affect cell shape and cellular barrier function (Prasain and Stevens 2009; Bogatcheva and Verin 2008).
To examine the mechanism underlying the effects of candesartan on endothelial cells during OGD/reoxygenation, we determined cell viability and the production of free radicals. Increased free radical production during ischemic conditions could increase endothelial permeability (Kaur and Ling 2008; Fraser 2011). Nitrate/nitrite plays several roles in cellular physiology, including as a free radical, endothelium-derived relaxing factor, and a neurotransmitter (Garthwaite 2008; Kvietys and Granger 2011). Nitrate/nitrite levels in the RBEC culture medium under OGD/reoxygenation were significantly higher than under normoxia. Although candesartan treatment (10 nM) tended to suppress nitrate/nitrite levels during OGD/reoxygenation, significant changes were not detected. Similarly, candesartan did not affect the production of ROS. Considering that reports have indicated that candesartan reduces protein nitration induced by peroxynitrite in an in vitro model (Soliman et al. 2014), a further study to examine protein oxidation levels during OGD/reoxygenation is necessary. In contrast, candesartan improved OGD/reoxygenation-induced cell damage. Thus, our data indicate that candesartan may maintain barrier integrity in primary RBEC cultures under hypoxia-reperfusion conditions by improving endothelial cell viability.
We could not detect an effect of candesartan on barrier properties under normoxic conditions, whereas candesartan improved barrier properties under OGD/reoxygenation. These results suggest that OGD/reoxygenation affects the Ang II systems in endothelial cells, possibly through increased production of Ang II or receptor modulation. We performed an enzyme immunometric assay (EIA) for Ang II to determine Ang II levels in the culture medium. Ang II levels were very low, near the detection limit of 5 pg/ml, and we were unable to detect significant differences between normoxia and OGD/reoxygenation (data not shown). Accumulated studies indicate that AT1 receptor stimulation by Ang II induces deleterious effects in cardiovascular disease, whereas the protective effects are mediated through the AT2 receptor. We also attempted to detect AT1/AT2 receptor levels under OGD/reoxygenation by immunoblotting. However, in agreement with Benicky et al. (2012) and Hafko et al. (2013), our antibodies were not capable of detecting these receptors. As Fleegal-DeMotta et al. (2008) indicated that AT2 receptor blockade does not affect BBB hyperpermeability induced by Ang II treatment, the role of AT2 receptors appears to be limited.
Recent reports indicate that several receptors, such as adrenergic, histamine, serotonin, dopamine, and angiotensin, exhibit constitutive activity in the absence of agonist stimulation (Milligan 2003; Khilnani and Khilnani 2011). Antagonists inhibit agonist-induced responses, but they do not have the ability to affect agonist-independent constitutive receptor activity. In contrast, inverse agonist binding to constitutively active receptors reduces their activity (Bond and Ijzerman 2006).
Some AT1 receptor blockers appear to act as inverse agonist. Candesartan is considered to be an inverse agonist and is different from other ARBs (Yasuda et al. 2008). Even in the absence of Ang II, AT1 receptors can be activated by mechanical stimulation, which would not be affected by AT1 receptor antagonist, but could be affected by inverse agonists such as candesartan (Zou et al. 2004). The effects of an inverse agonist on an RBEC monolayer model are shown here, where it is thought that candesartan, in the absence of Ang II, controlled downstream signaling of the AT1 receptor. Additionally, another potential mechanism of candesartan-mediated BBB protection may be due to activation of the AT1 receptor during OGD/reoxygenation, which is blocked by candesartan binding to the activated AT1 receptor.
In summary, candesartan inhibits increased paracellular BBB transport under OGD/reoxygenation conditions. This mechanism may lead to improved cell survival, with candesartan treatment representing an important clinical tool for patients with cerebral ischemia.
References
Abbott NJ (2005) Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol 25:5–23
Benicky J, Hafko R, Sanchez-Lemus E, Aguilera G, Saavedra JM (2012) Six commercially available angiotensin II AT1 receptor antibodies are non-specific. Cell Mol Neurobiol 32:1353–1365
Bogatcheva NV, Verin AD (2008) The role of cytoskeleton in the regulation of vascular endothelial barrier function. Microvasc Res 76:202–207
Bond RA, Ijzerman AP (2006) Recent developments in constitutive receptor activity and inverse agonism, and their potential for GPCR drug discovery. Trends Pharmacol Sci 27:92–96
Cohn JN, Tognoni G, Valsartan Heart Failure Trial Investigators (2001) A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 345:1667–1675
Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H, LIFE Study Group (2002) Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359:995–1003
Deli MA, Ábrahám CS, Kataoka Y, Niwa M (2005) Permeability studies on in vitro blood-brain barrier models: physiology, pathology and pharmacology. Cell Mol Neurobiol 25:59–127
Dirnagl U (2012) Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci 1268:21–25
Edvinsson LI, Povlsen GK (2011) Vascular plasticity in cerebrovascular disorders. J Cereb Blood Flow Metab 31:1554–1571
Engelhardt B (2003) Development of the blood–brain barrier. Cell Tissue Res 314:119–129
Faure S, Bureau A, Oudart N, Javellaud J, Fournier A, Achard JM (2008) Protective effect of candesartan in experimental ischemic stroke in the rat mediated by AT2 and AT4 receptors. J Hypertens 26:2008–2015
Fleegal-DeMotta MA, Doghu S, Banks WA (2008) Angiotensin II modulates BBB permeability via activation of the AT(1) receptor in brain endothelial cells. J Cereb Blood Flow Metab 29:640–647
Fraser PA (2011) The role of free radical generation in increasing cerebrovascular permeability. Free Radic Biol Med 51:967–977
Garthwaite J (2008) Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci 27:2783–2802
Hafko R, Villapol S, Nostramo R, Symes A, Sabban EL, Inagami T, Saavedra JM (2013) Commercially available angiotensin II At2 receptor antibodies are nonspecific. PLoS ONE 8:e69234
Hiu T, Nakagawa S, Hayashi K, Kitagawa N, Tsutsumi K, Kawakubo J, Honda M, Suyama K, Nagata I, Niwa M (2008) Tissue plasminogen activator enhances the hypoxia/reoxygenation-induced impairment of the blood-brain barrier in a primary culture of rat brain endothelial cells. Cell Mol Neurobiol 28:1139–1146
Horai S, Nakagawa S, Tanaka K, Morofuji Y, Couraud PO, Deli MA, Ozawa H, Niwa M (2013) Cilostazol strengthens barrier integrity in brain endothelial cells. Cell Mol Neurobiol 33:291–307
Ishizaki T, Chiba H, Kojima T, Fujibe M, Soma T, Miyajima H, Nagasawa K, Wada I, Sawada N (2003) Cyclic AMP induces phsophorylation of claudin-5 gene in blood-brain barrier endothelial cells via protein-kinase A-dependent and -independent pathway. Exp Cell Res 290:275–288
Kataoka H, Murakami R, Numaguchi Y, Okumura K, Murohara T (2010) Angiotensin II type 1 receptor blockers prevent tumor necrosis factor-alpha-mediated endothelial nitric oxide synthase reduction and superoxide production in human umbilical vein endothelial cells. Eur J Pharmacol 636:36–41
Kaur C, Ling EA (2008) Blood brain barrier in hypoxic-ischemic conditions. Curr Neurovasc Res 5:71–81
Khilnani G, Khilnani AK (2011) Inverse agonism and its therapeutic significance. Indian J Pharmacol 43:492–501
Kozak A, Ergul A, El-Remessy AB, Johnson MH, Machado LS, Elewa HF, Abdelsaid M, Wiley DC, Fagan SC (2009) Candesartan augments ischemia-induced proangiogenic state and results in sustained improvement after stroke. Stroke 40:1870–1876
Kvietys PR, Granger DN (2011) Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic Biol Med 52:556–592
Lindholm LH, Ibsen H, Dahlof B, Devereux RB, Beevers G, de Faire U, Fyhrquist F, Julius S, Kjeldsen SE, Kristiansson K, Lederballe-Pedersen O, Nieminen MS, Omvik P, Oparil S, Wedel H, Aurup P, Edelman J, Snapinn S (2002) Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet 359:1004–1010
Lu Q, Zhu YZ, Wong PT (2005) Neuroprotective effects of candesartan against cerebral ischemia in spontaneously hypertensive rats. NeuroReport 16:1963–1967
Milligan G (2003) Constitutive activity and inverse agonists of G protein-coupled receptors: a current perspective. Mol Pharmacol 64:1271–1276
Nakagawa S, Deli MA, Nakao S, Honda M, Hayashi K, Nakaoke R, Kataoka Y, Niwa M (2007) Pericytes from brain microvessels strenghthen the barrier integrity in primary cultures of rat brain endothelial cells. Cell Mol Neurobiol 27:687–694
Nakagawa S, Deli MA, Kawaguchi H, Shimizudani T, Shimono T, Kittel A, Tanaka K, Niwa M (2009) A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem Int 54:253–263
Ogihara T, Nakao K, Fukui T, Fukiyama K, Ueshima K, Oba K, Sato T, Saruta T, Candesartan Antihypertensive Survival Evaluation in Japan Trial Group (2008) Effects of candesartan compared with amlodipine in hypertensive patients with high cardiovascular risks: candesartan antihypertensive survival evaluation in Japan trial. Hypertension 51:393–398
Panahpour H, Nekooeian AA, Dehghani GA (2014) Candesartan attenuates ischemic brain edema and protects the blood-brain barrier integrity from ischemia/reperfusion injury in rats. Iran Biomed J 18:232–238
Pelisch N, Hosomi N, Ueno M, Nakano D, Hitomi H, Mogi M, Shimada K, Kobori H, Horiuchi M, Sakamoto H, Matsumoto M, Kohno M, Nishiyama A (2011) Blockade of AT1 receptors protects the blood-brain barrier and improves cognition in Dahl salt-sensitive hypertensive rats. Am J Hypertens 24:362–368
Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, Yusuf S, Pocock S (2003) Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-overall programme. Lancet 362:759–766
Prasain N, Stevens T (2009) The actin cytoskeleton in endothelial cell phenotypes. Microvasc Res 77:53–63
Saavedra JM (2012) Angiotensin II AT1 receptor blockers as treatments for inflammatory brain disorders. Clin Sci 123:567–590
Schmieder RE, Hilgers KF, Schlaich MP, Schmidt BM (2007) Renin-angiotensin system and cardiovascular risk. Lancet 369:1208–1219
Schrader J, Lüders S, Kulschewski A, Berger J, Zidek W, Treib J, Einhäupl K, Diener HC, Dominiak P, Acute Candesartan Cilexetil Therapy in Stroke Survivors Study Group (2003) The ACCESS Study: evaluation of Acute Candesartan Cilexetil Therapy in Stroke Survivors. Stroke 34:1699–1703
Skoog I, Lithell H, Hansson L, Elmfeldt D, Hofman A, Olofsson B, Trenkwalder P, Zanchetti A, SCOPE Study Group (2005) Effect of baseline cognitive function and antihypertensive treatment on cognitive and cardiovascular outcomes: study on COgnition and Prognosis in the Elderly (SCOPE). Am J Hypertens 18:1052–1059
Soliman S, Ishrat T, Pillai A, Somanath PR, Ergul A, El-Remessy AB, Fagan SC (2014) Candesartan induces a prolonged proangiogenic effect and augments endothelium-mediated neuroprotection after oxygen and glucose deprivation: role of vascular endothelial growth factors A and B. J Pharmacol Exp Ther 349:444–457
Wosik K, Cayrol R, Dodelet-Devillers A, Berthelet F, Bernard M, Moumdjian R, Bouthillier A, Reudelhuber TL, Prat A (2007) Angiotensin II controls occludin function and is required for blood brain barrier maintenance: relevance to multiple sclerosis. J Neurosci 27:9032–9042
Yamada K, Nabeshima T (1997) Simultaneous measurement of nitrite and nitrate levels as indices of nitric oxide release in the cerebellum of conscious rats. J Neurochem 68:1234–1243
Yamamoto M, Ramirez SH, Sato S, Kiyota T, Cerny RL, Kaibuchi K, Persidsky Y, Ikezu T (2008) Phosphorylation of caludin-5 and occluding by Rho kinase in brain endothelial cells. Am J Pathol 172:521–533
Yasuda N, Akazawa H, Qin Y, Zou Y, Komuro I (2008) A novel mechanism of mechanical stress-induced angiotensin II type 1-receptor activation without the involvement of angiotensin II. Naunyn Schmiedebergs Arch Pharmacol 377:393–399
Youdim KA, Avdeef A, Abbott NJ (2003) In vitro trans-monolayer permeability calculations: often forgotten assumptions. Drug Discov Today 8:997–1003
Zaman MA, Oparil S, Calhoun DA (2002) Drugs targeting the renin-angiotensin-aldosterone system. Nat Rev Drug Discov 1:621–636
Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T (2010) Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 171:852–858
Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6:499–506
Acknowledgments
Candesartan was a generous gift from Takeda Pharmaceutical Co., Ltd., Japan. This work was supported in part by a Grant-in-Aid for Young Scientists (B) (24790258) from the Japan Society for the Promotion of Science (JSPS). We wish to thank Yasuko Yamashita, Takanori Shimono, Narumi Yamada, and Rie Tatsumi for their critical reviews of the manuscript and outstanding professional guidance.
Conflict of interest
MN is an unsalaried president of PharmaCo-Cell Co. Ltd. (Nagasaki, Japan). All other authors have no competing interests. PharmaCo-Cell Co. Ltd., a University spin-off company, is a company that does business in the field of BBB research. This research does not present any financial conflicts.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
So, G., Nakagawa, S., Morofuji, Y. et al. Candesartan Improves Ischemia-Induced Impairment of the Blood–Brain Barrier In Vitro. Cell Mol Neurobiol 35, 563–572 (2015). https://doi.org/10.1007/s10571-014-0152-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10571-014-0152-8