
Abstract
This research focuses on the application of nanocellulose-stabilized silver nanoparticles (AgNPs) as a colorimetric sensing probe for mercury (II) detection. For this purpose, cellulose nanocrystals (CNCs) and cellulose nanofibers (CNFs) were employed with dual functions: a stabilizing agent and a dispersing matrix. The chemical and structural properties of the samples were analysed by various techniques such as XRD, FTIR, TEM and UV–Vis Spectroscopy. Due to different morphologies and surface functional groups in each type of nanocellulose, their effects on the AgNP formation as well as their response against mercury (II) ions were studied. It was discovered that the AgNPs stabilized by both types of nanocellulose can provide high sensitivity and selectivity towards mercury (II) ions. However, some interference caused by iron (III) ions was observed. This interference could be significantly reduced if the CNFs were employed during the AgNP preparation. Therefore, the advantage of using the CNFs as a stabilizing agent as well as a dispersing matrix will be discussed in detail. Furthermore, the possibility of fabrication into solid-state platforms (nanopapers) and response to mercury (II) ions will be demonstrated.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nowadays, mercury contamination remains a serious problem in the world. Besides the impact to ecosystems, mercury poisoning can cause adverse health effects to humans which can be transferred to future generations (Gad and Pham 2014). Mercury, naturally found in the earth's crust, can be liberated to the atmosphere by different ways such as burning of coal-fired power plants, burning of wastes, or released during industrial processes. Harmfully, mercury can enter the food chain and subsequently accumulate in living organisms due to biomagnification (Gworek et al. 2020; Driscoll et al. 2013; Li and Tse 2015). In humans, exposure to mercury can cause severe damage to several organs, i.e., brain, thyroid, breast, myocardium, muscles, skin, pancreas, liver, and kidneys (Andreoli and Sprovieri 2017; Dasharathy et al. 2022; Engström et al. 2013). Mercury circulates through the environment (air, land and water) in different forms including elemental mercury (Hg0), inorganic mercury (Hg2+ and Hg+) and organic mercury (CH3Hg+ and C2H5Hg+) (Gad and Pham 2014). Since mercury can cause irreversible damage to human health even at low exposure, monitoring of mercury concentration in different resources becomes an important issue for scientists.
Currently, there are lots of technologies applied to mercury detection such as atomic absorption spectrometry (AAS), inductively coupled plasma mass spectrometry (ICP-MS), X-ray fluorescence spectrometry (XRF), etc. However, these techniques must be performed in a laboratory and require expensive equipment. For this reason, several kinds of sensing systems were developed, especially for the detection of mercury (II) ions, which is one of the most stable forms (Dai et al. 2021). Among numerous approaches, metal nanoparticle-based sensors have drawn great attention due to their promising properties, sensitivity, and selectivity, in particular, gold nanoparticles (AuNPs) and silver nanoparticles (AgNPs) (Gul et al. 2022). Different modes of detection were employed to attain a very high limit of detection (LOD) including electrochemical, colorimetric, and fluorescent (Logan et al. 2021; Crockett et al. 2021; Wang et al. 2022; Narouei et al. 2021; Lin et al. 2022; Vyas et al. 2019; Qing et al. 2018; Pomal et al. 2021; Hashemi et al. 2022; Bhattacharjee et al. 2018). However, colorimetric analysis has more advantages in terms of simplicity, versatility, and cost-effectiveness. Therefore, this approach was selected in our study, and we focused on the utilization of AgNPs as colorimetric sensing probes. The strategy is based on the redox reaction between Ag(0) and Hg(II) ions which can induce the formation of solid Ag-Hg amalgam and finally lead to a change in color as well as their absorption spectra. The localized surface plasmon resonance (LSPR) of the AgNPs will allow us to observe their color change even with naked eyes. Depending on the fabrication methods and the types of stabilizing agent employed, different limits of detection can be achieved ranging from a few nanomolar to micromolar levels (Zangeneh Kamali et al. 2016; Vasileva et al. 2017; Ismail et al. 2019; Ghosh and Mondal 2020; Faghiri and Ghorbani 2019; Budlayan et al. 2022; Balasurya et al. 2020). Despite the satisfactory results, most of these sensors are in liquid phase, thereby, they still require proper storage to maintain high sensing accuracy.
To our knowledge, no research has been carried out using nanocellulose as a stabilizing agent in the preparation of AgNP-based sensors. However, this polysaccharide polymer may offer great benefit. Firstly, it consists of several glucose units linked by glycosidic bonds which provide a large number of hydroxyl groups. Thus, it can be either chemically modified or grafted with other compounds in order to incorporate some specific functional groups on the surface. (Klemm et al. 2018; Lin et al. 2012) Furthermore, this plant-based material is promising to be developed into paper-based platform. (Marquez and Morales-Narváez 2019; Golmohammadi et al. 2017; Morales-Narváez et al. 2015; Lokanathan et al. 2015) Secondly, the surface of nanocellulose extracted by chemical routes are negatively charged, thus, they are suitable for AgNP stabilization. Besides, in sensing applications, this condition will favor the attraction of target metal ions close to the sensing probes leading to sensitive and selective detection. Hence, in our present work, we have chosen two types of nanocellulose obtained by different procedures; (i) cellulose nanocrystals (CNCs) synthesized by acid hydrolysis providing sulfate functional groups (− OSO3‾) (Song et al. 2019) and (ii) cellulose nanofibers (CNFs) prepared by TEMPO-mediated oxidation offering carboxylate functional groups (− COO‾) on the surface (Ovalle-Serrano et al. 2020). As the CNCs and the CNFs have different structures and surface functional groups, their influence on the synthesis of AgNPs as well as on sensing of mercury (II) ions will be investigated. By serving as a stabilizing agent and a dispersing matrix, the efficiency of different nanocellulose in mercury (II) detection will be compared. We will also discuss the interference triggered by the presence of iron (III) ions for this sensing system. Finally, some examples of solid-state nanopapers will be presented and their sensing ability toward mercury (II) will be explored.
Materials and methods
Materials
Silver nitrate (AgNO3) and microcrystalline cellulose (MCC) derived from cotton linters were obtained from Merck, Germany. Sulfuric acid (H2SO4), hydrochloric acid (HCl), sodium hydroxide (NaOH), sodium bromide (NaBr), sodium hypochorite (NaClO) and hydrogen peroxide (H2O2) were purchased from Qrëc, New Zealand. Tetramethylpiperidine-1-oxyl (TEMPO) and sodium borohydride (NaBH4) were supplied by Sigma-Aldrich, France. Water-soluble metal salts such as mercuric chloride (HgCl2), nickel chloride (NiCl2), lead chloride (PbCl2), cobalt chloride (CoCl2), strontium chloride (SrCl2), cadmium chloride (CdCl2), sodium chloride (NaCl), potassium (KCl), zinc sulfate (ZnSO4), copper sulfate (CuSO4), manganese sulfate (MnSO4), magnesium sulfate (MgSO4), calcium sulfate (CaSO4) and iron nitrate Fe(NO3)3 were purchased from Ajax Finechem, New Zealand. All chemicals were used as received.
Preparation of nanocellulose
Cellulose nanocrystals (CNCs)
To prepare the CNCs, 3 g of MCC powder (75 – 250 μm) were slowly added to 30 mL of 60% sulfuric acid, placed in an ice bath. The mixture was stirred vigorously for 5 min. After that, it was brought to room temperature and kept stirring for 30 min (Song et al. 2019). The hydrolysis was stopped by adding 300 mL of cold DI water into the solution. The residual was eliminated by using the centrifugation at 12,000 rpm (14,489 × g), 10 °C, for 15 min. Next, the obtained CNCs were rinsed 3 times and dialyzed against ultra-pure water for 1 week. Finally, the concentration of CNC suspension was determined by using the TGA technique.
Cellulose nanofibers (CNFs)
The CNFs were prepared by TEMPO-oxidation method (Ovalle-Serrano et al. 2020) using also the MCC powder as starting material. The procedure began by dispersing 3 g of MCC in 100 mL of DI water. Later, 16 mg of TEMPO and 100 mg of sodium bromide were added to the mixture. With stirring, 22.4 mL of 5% sodium hypochlorite solution was added dropwise to initiate the TEMPO-oxidation. The reaction was carried out at room temperature for about 3 h while the pH was maintained between 10–11. The reaction was quenched by adding 5 mL of ethanol into the mixture and neutralized with 0.5 M HCl solution. The obtained CNFs were cleansed 3 times using centrifugation at 12,000 rpm (14,489 × g), followed by dialysis against ultra-pure water for 1 week. Finally, the concentration of the CNFs in suspension was determined.
Homogenization of the nanocellulose suspensions
The concentrations of CNCs and CNFs were adjusted to 1wt% before being submitted to mechanical disintegration using a 750-W ultrasonicator with a ½” diameter probe. The process was performed at 40% amplitude with 1:1 pulse until a clear transparent suspension was obtained.
Synthesis of silver nanoparticles
To synthesize silver nanoparticles (AgNPs), 0.5 mL of the 1wt% CNCs or CNFs was introduced to 20 mL of 1 mM AgNO3 and stirred for 5 min. Then, the mixture was poured into 30 mL of 1 mM NaBH4 placed in an iced bath under darkness and kept stirring for 40 min. Finally, the AgNPs synthesized in 0.01wt% of CNCs or CNFs were denoted as AgNP@CNC and AgNP@CNF.
Colorimetric sensing of mercury (II) ions
The AgNP@CNC and AgNP@CNF were utilized to prepare different solutions for analyses. To understand the effect of nanocellulose types on the sensing property of the AgNPs and the effect of nanocellulose used in excess as dispersing matrix, our experiments were separated into 3 parts (Experiments 1–3) as presented in Table 1. For each measurement, pure nanocellulose (1wt% CNCs or 1wt% CNFs) were added to the AgNP solution and the total volume was adjusted to 2 mL by ultra-pure water. Their final concentrations in wt% are displayed in Table 1. Afterwards, 0.50 mL of Hg (II) solution with concentrations in the range of 1–1000 μM was added, and the sensing response was examined. Moreover, to study the selectivity of our sensors, different metal ions were employed (i.e., Na(I), K(I), Ca(II), Mg(II), Sr(II), Pb(II), Mn(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II) and Fe(III)), and their sensing response will be compared with that of Hg(II).
Preparation of the AgNP nanopapers
The AgNP nanopapers were prepared by simply drying the solution according to experiment 3 in a clear plastic mold (Ø 3.3 cm), with double the amount. The drying process was carried out in the dark under ambient conditions. The resultant yellow transparent nanopapers were eventually maintained in the mould and stored in a desiccator before usage.
Characterization Techniques
A thermogravimetric analyser (Perkin-Elmer TGA8000) was used to determine the concentration of nanocellulose in aqueous suspension. The sample was dropped on an alumina TGA pan and heated from room temperature to 120 °C with a heating rate of 5 °C min−1 under nitrogen atmosphere. The mass loss due to water removal was measured for five samples and the average value was applied for calculation. The crystalline phase of nanocellulose was examined by a Bruker D8 Advance diffractometer operated at 40 kV/40 mA using CuKα radiation at a wavelength of 0.154 nm. For this analytical technique, the CNC/CNF powders obtained by freeze drying, were utilized. The XRD data were collected over an angular range of 10–60° at room temperature. Fourier transform infrared spectroscopy (FTIR) analysis of the CNCs and CNFs was performed by a Perkin-Elmer UATR-2 FT-IR spectrophotometer using a transmittance mode in the range of 4000–500 cm−1 with a resolution of 1 cm−1. A transmission electron microscope (TEM-Hitachi HT7700) was employed to explore the morphologies of the samples. The AgNP suspension was dropped onto a holey carbon coated TEM grid, which was then left to dry in air. The observations were conducted in the magnification range from 10000 × to 50,000 × at 80 kV acceleration voltage. The particle size distribution was evaluated using Image-J analysis software. The surface plasmon resonance (SPR) of the AgNPs within each sample was monitored by a UV–Visible spectrophotometer (Lamda365, Perkin Elmer instrument) in the range of 300–800 nm. The surface charge of the samples was also measured by a Zetasizer instrument (Nano series, Malvern Instruments Ltd., UK).
Results & discussion
Characterizations of nanocellulose
The XRD patterns of the microcrystalline cellulose (MCC) and the nanocellulose (CNC/CNF) are illustrated in Fig. 1a. The diffraction peaks at about 14.8°, 16.3°, 22.4° and 34.5°2θ related to (1–10), (110), (200) and (004) Miller indices were observed for all samples. These peaks matched well with the database of cellulose Iβ (French 2014). One additional peak was also observed at 12.2° for the CNCs indicating that the phase transformation from cellulose Iβ to II occurred. However, the high intensity of the major peaks confirmed the predominance of cellulose Iβ in the sample. Considering the preparation method of the CNCs, the formation of cellulose II might be caused by the regeneration of cellulosic chains in some regions after being exposed to acid hydrolysis (Sèbe et al. 2012). The crystallinity index (CrI) of the samples was calculated by using Segal formula (Segal et al. 1959):
where I200 was the height of the 200 peak (at 2θ = 22.4°) and Iam was the height of the minimum between the 110 and the 200 peaks (at 2θ = 18.6°). A straight line connecting the two points at 10° and 60° was applied as baseline and used for measuring the I200 and Iam, as illustrated in Fig. 1a. The obtained CrI values for the MCC, CNFs and CNCs were 89.4%, 88.3%, and 80.6%, respectively. It might be stated that the lowering of the CrI value in the CNCs was caused by the presence of some intensity from the 110 peak of cellulose II. The Iam position was therefore shifted to the lower 2θ with a little increase in intensity. The FT-IR analyses of the MCC, CNCs and CNFs were performed as depicted in Fig. 1b. The peaks appeared around 3320 cm−1 and 2895 cm−1 corresponded to the O–H and C-H stretching vibrations in polysaccharides. The peaks related to O–H bending in celluloses and in adsorbed water were observed at 1646 cm−1 for the CNCs and 1598 cm−1 for the CNFs. The stronger intensity for the latter may be due to the overlapping of the peaks between the O–H bending and the carboxylate groups produced by TEMPO-oxidation (Yang et al. 2017). The antisymmetric bridge stretching of C–O–C groups was also found at 1160 cm−1 (Foster et al. 2018). The peaks at 1109, 1055 and 1035 cm−1 could be attributed to C-O stretching at C2, C3 and C6 of the pyranose rings, respectively (Foster et al. 2018). The C-H rocking vibration in celluloses was likewise detected at around 899 cm−1 (Li et al. 2009).

XRD (a) and FTIR (b) spectra of microcrystalline cellulose (MCC), cellulose nanocrystals (CNCs) and cellulose nanofibers (CNFs)
Characterizations of silver nanoparticles
In this work, the AgNPs were synthesized using the CNCs or the CNFs as a stabilizer/dispersant. The resulting solutions exhibited a clear yellow-colour indicating that the AgNPs were homogenously dispersed. The solutions, namely AgNP@CNC and AgNP@CNF, were then observed using a transmission electron microscope (TEM) without any dilution, as shown in Fig. 2(a-d). The histograms in the insets of Fig. 2a and 2c revealed that the AgNPs had a narrow size distribution. According to statistical analysis based on 200 nanoparticles, the average diameters were 7.9 ± 0.3 nm for AgNP@CNC and 15.6 ± 0.3 nm for AgNP@CNF. Although smaller AgNPs were obtained in the case of CNC, they were slightly aggregated. This difference might be related to a faster nucleation rate in the CNC medium which yielded small particles with high surface energy, whereas the slower nucleation rate in the CNF condition was favourable to the formation of larger particles. Moreover, most of the formed AgNPs were found on nanocellulose surfaces, especially for the CNCs, which by themselves were slightly aggregated. The presence of the CNCs and the CNFs in each sample can be well visualized at high image contrast (grey colour distinguished from the background). According to the zeta potential measurements (Fig.S1(a and c)), the surface charge of the AgNP@CNC (− 18 mV) was much lower than that of the AgNP@CNF (− 39 mV). Hence, their particles would aggregate more easily due to the decrease of electrostatic repulsion (Uddin et al. 2014; Lokanathan et al. 2014). This hypothesis is consistent with the hydrodynamic size measured in our samples (106.6 nm for AgNP@CNC and 73.71 nm for AgNP@CNF with the PDI values of 0.580 and 0.566, respectively (Fig.S1(b and d)). The lower stability of the NPs in CNC medium is thus the reason for the aggregation observed in TEM images.

TEM micrographs and size distribution of AgNPs synthesized by using (a—b) the CNC suspension (AgNP@CNC), and (c—d) the CNF suspension (AgNP@CNF)
Sensing of mercury (II) ions in aqueous solution
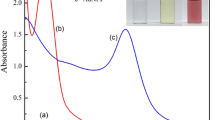
The colorimetric sensing of Hg(II) ions was carried out by monitoring the change in local SPR band of the AgNP@CNC and the AgNP@CNF solutions. With some addition of pure CNCs or CNFs, as described in Table 1, the sensing properties toward Hg(II) ions were explored. Figure 3(a, b, and c) illustrate the local SPR band of the AgNPs after interaction with the Hg(II) ions at different concentrations, i.e., 0, 1, 10, 20, 50, 100, 150, 250, 500, 750, 1000 µM. As the results in experiments 1–3 followed the same trend, only those of the AgNP@CNC and the AgNP@CNF with the addition of 0.75 mL of CNCs or 0.75 mL of CNFs were presented. It can be seen in Fig. 3 that, without interaction with Hg(II) ions, the AgNPs exhibited strong SPR peaks with the maxima (λmax) around 398 nm. A slight difference in FWHM was observed between the AgNP@CNC and the AgNP@CNF, indicating that larger particles were formed when using the CNFs as dispersants, in accordance with TEM observation. After the addition of Hg(II) ions, a decrease in intensity of the SPR band with a blueshift of the λmax was observed along with the decolorization from clear yellow to colourless. The colour change could even be observed with the naked eye after interaction with 100–250 µM of Hg(II) upon the sensing condition, and complete decolorization was achieved when the concentration was over 500 µM (see the photographs in Fig. 4). As is widely known in the literature, the shift or decrease of the SPR band originates from the electron transfer from metallic silver to mercury (II) ions, since the standard reduction potential of mercury (EºHg(II)/Hg(0) = + 0.85 V) is higher than that of silver (EºAg(I)/Ag(0) = + 0.80 V). Moreover, the Hg(0) metal produced by the redox reaction has a strong affinity toward silver. It can be adsorbed onto the surface of AgNPs, leading to the formation of Ag-Hg amalgams which destroy the SPR property of the AgNPs (Zangeneh Kamali et al. 2016).

UV–Visible absorbance of (a) AgNP@CNC/ 0.375 wt% CNCs, (b) AgNP@CNC/ 0.375 wt% CNFs and (c) AgNP@CNF/ 0.375 wt% CNFs, after addition of Hg(II) solution at different concentrations

Plot of absorbance ratio (A370/A398) vs Hg(II) concentration for (a and c) the AgNP@CNC and (e) the AgNP@CNF, mixed with some additional CNC or CNF suspensions presented in wt%; (b, d, and f) photographs showing the decolorization of the sensing system after interacting with Hg(II) ions
To understand the dependence of nanocellulose types on the sensitivity of AgNPs toward Hg(II) ions, the results from Experiments 1, 2 and 3 were presented by plotting the absorbance ratio (A370/A398) as a function of Hg(II) concentration (Fig. 4(a-f)). Note that, between 370 and 398 nm, a significant change in absorbance was detected. For Experiment 1, the AgNPs were synthesized using the CNCs as stabilizer and added up with different quantities of 1wt% CNCs before being submitted to Hg(II) detection, namely AgNP@CNC/CNC. As seen in Fig. 4a, the A370/A398 ratio increased when the concentration of Hg(II) increased. This signifies that the absorbance curve was changing its maximum (blueshift) along with the decrease in intensity. Moreover, the ratio increased sharply for low Hg(II) concentrations and slowed down after 50 µM before reaching a plateau at around 500 µM. In fact, this turning point revealed that strong decolorization was taking place and the absorbance ratio would reach its highest value when the AgNPs were completely decolorized. This result was in accordance with the photographs presented in Fig. 4b. Although the same trends were obtained in this case, we observed (with the naked eye) a stronger decolorization in the range of 50–250 µM of Hg(II), when some quantities of pure CNCs were added into the sensing solutions. Since, initially, the AgNP@CNCs were slightly agglomerated as seen in Fig. 2(a-b) for TEM images and the CNCs by themselves had some drawbacks in terms of stability, a self-agglomeration could easily occur, therefore the solutions became fainter. This phenomenon might also be the reason for the instability of the A370/A398 curves in Experiment 1. To reduce the agglomeration problem, we also tried to utilize the CNFs as additional dispersant instead of the CNCs (Experiment 2). The results for AgNP@CNC/CNF are shown in Fig. 4c and 4d. In this system, the agglomeration was still observable. The fluctuation of the A370/A398 curves still existed and no relationship between the data and the quantity of CNFs was found. For Experiment 3, the AgNPs were synthesized using the CNFs instead of the CNCs and also applied as additional dispersants for Hg(II) detection. Surprisingly, with this condition, satisfactory results could be obtained (Fig. 4e and 4f). As seen in the photographs in Fig. 4f, the decolorization of AgNP@CNF/CNF occurred gradually when the concentration of Hg(II) ions increased. Furthermore, for all quantities of the CNFs added up, similar A370/A398 curves were obtained (Fig. 4e). This indicates that only the CNFs employed during the synthesis of the AgNP@CNF were sufficient to provide a good dispersion and extra quantities had a very low impact on the sensing property of the AgNPs. However, as previously discussed, this advantage might be related to the high surface charge of the CNFs, which provides adequate repulsion among the AgNPs. Furthermore, their steric hinderance would be beneficial to AgNP dispersibility and lead to efficient Hg(II) ion penetration.
The limits of detection (LOD) were also calculated in this work by considering the absorbance ratio curves in the range of 0–50 μM Hg (II) concentrations. A linear relationship obtained in this area gave average LOD values of around 3.44, 4.07 and 3.99 μM for the AgNP@CNC/CNC, the AgNP@CNC/CNF, and the AgNP@CNF/CNF, respectively. Despite, the comparable LOD values, the results from the absorbance ratio curves demonstrated that the AgNP@CNF/CNF were the most efficient because less variation of the data was obtained.
Selectivity and interference studies
The selectivity study was performed by adding different metal ions into the as prepared sensors, including Na(I), K(I), Ca(II), Mg(II), Sr(II), Pb(II), Mn(II), Co(II), Ni(II), Cu(II), Zn(II), Cd(II), Hg(II) and Fe(III). Their concentrations were fixed at 1 mM and the change in the absorbance ratio was evaluated as shown in Fig. 5(a and b). From the results, no response to any metal ions was found except for the Hg(II) and Fe(III) ions. The photographs of the AgNP solutions after interacting with different metal ions are presented in the insets of Fig. 5. It can be seen that with the presence of Hg(II) ions, the AgNPs decolorized completely due to the higher standard reduction potential of Hg(II) ions which converted the Ag(0) to Ag(I) ions. However, in the case of Fe(III) ions, the decolorization also occurred despite the inadequate redox condition (EºFe(III)/Fe(II) = + 0.77 V).

Selectivity study against various metal ions; photographs and the change of absorbance ratio (a) for the AgNP@CNC/CNC and (b) for the AgNP@CNF/CNF
Indeed, the response of the AgNPs toward Fe(III) ions has already been observed by several research groups, especially for biogenic AgNPs. Most explanations refer to the exceptional redox process or the formation of a Fe(III) chelating complex leading to agglomeration and decolorization of the AgNPs (Gao et al. 2015; Kumar et al. 2017; Ghosh et al. 2018; Chandraker et al. 2019; Trang et al. 2021; Doan et al. 2022). Recently, Maity et al. (Maity et al. 2018) also proposed a novel mechanism for the oxidation of the AgNPs in organometallic media. It was suggested that beyond the modulation of the Fe(III)/Fe(II) reduction potential, the AgNPs oxidation could be reinforced by the formation of a stable Fe(II)-complex which finally caused the AgNP decolorization. Based on the results obtained in our work, we have paid attention to the formation of chelating complexes because a strong decolorization was observed for the AgNP@CNC/CNC, but not for the AgNP@CNF/CNF (Fig. 5a and b). As discussed in the previous section, the CNFs provided better dispersion for the AgNPs; thus, the agglomeration caused by the formation of Fe(III) chelating complexes might be inhibited due to the high negative surface charge of the CNFs which generated a strong repulsion between the particles in solution. To confirm this hypothesis, a complementary experiment was performed using the AgNP@CNF/CNF as a sensing probe and the concentration of the Fe(III) ions was varied between 0 – 10 mM. The response of the AgNP@CNF/CNF toward Fe(III) ions was displayed in the supplementary information (Fig. S2). It can be seen in Fig.S2(a) that the SPR bands of the AgNP@CNF/CNF were nearly constant after reacting with the Fe(III) ions for concentrations below 500 μM. This implied that the Fe(III) ions had very low influence in this concentration range. Nevertheless, at higher concentrations, the agglomeration effect could happen and at this point, the SPR band would dramatically change. As shown in Fig.S2(a), the SPR band decreased sharply at 10 mM of Fe(III), and some aggregates could be clearly observed with the naked eye (photograph in Fig.S2(b)). To demonstrate that the CNFs can retard the agglomeration, a similar experiment was carried out, but without the addition of extra CNF suspensions (Fig. S3). Here, at only 1 mM of Fe(III), large aggregates were significantly formed, and the remaining solution became transparent. This strongly confirmed the protecting ability of the CNFs against agglomeration. According to the overall results, we assumed that the CNFs were favorable to be employed as dispersants in this sensing system because the agglomeration effect can be greatly delayed.
Although the interference induced by the Fe(III) ions was unavoidable in the solution phase, the activity of the Hg(II) ions was still predominant. As shown in Fig. 6, when the Hg(II) ions were mixed with different metal ions, all solutions became completely colorless, except when mixing with the Fe(III) ions. In this case, the pale-yellow colour of the solution might not be generated from the SPR of the AgNPs, but it should be the original colour of the Fe(III) solution. Therefore, the origin of decolorization still pointed to the oxidation of AgNPs involved by the Hg(II) ions.

Sensing property of (a) the AgNP@CNC/CNC and (b) the AgNP@CNF/CNF toward Hg(II) ions in the presence of other metal ions
Sensing performance of the AgNP@CNF/CNF nanopapers
To demonstrate that our sensors are promising to be developed into solid-state platforms, some examples of AgNP based nanopaper were fabricated. The AgNP@CNF/CNF solutions were employed and transformed into nanopapers by simply air-drying in the dark. Herein, the protocol of Experiment 3 was employed with double the amount of solution. The resulting samples were labeled as CNF-0.0, CNF-0.5, CNF-1.0, CNF-1.5, CNF-2.0 or CNF-2.5, depending on the volume of 1 wt% CNFs added. The photographs of the AgNP@CNF/CNF nanopapers obtained after the drying process were shown in Fig.S4. It was found that the CNFs played an important role in the production of nanopaper. An adequate quantity of the CNFs was required in order to obtain a well-defined nanopaper. Finally, the CNF-1.5, CNF-2.0 and CNF-2.5 were selected for sensing performance tests and the results are shown in Fig. 7. It can be seen that all samples reacted well with the Hg(II) solutions. An evident change could be noticed from 50 μM of Hg(II) and complete decolourization could be achieved at 750 μM. We realized also that the as prepared nanopapers provided excellent mechanical strength; after soaking in an Hg(II) solution, their original form could be maintained. An example of a post-reacted nanopaper is also presented in Fig.S5. Finally, the overall results revealed that the AgNP@CNF/CNF nanopaper could be an efficient solid-state nanosensor for the colorimetric detection of Hg(II) ions and it would be particularly convenient for on-site applications.

Sensing performance tests of the AgNP@CNF nanopapers against Hg(II) ions at different concentrations
Conclusion
The AgNPs stabilized by nanocellulose (AgNP@CNC and AgNP@CNF) were applied as colorimetric sensing probes for Hg(II) detection. A sensitive detection against Hg(II) ions was obtained giving the LOD of around 3.8 μM. The change in colour from yellow to colourless can be observed even with the naked eye. When additional nanocellulose was introduced into the sensing solutions, its performance could be maintained. This was advantageous since the nanocellulose would serve as a matrix during nanopaper fabrication; the AgNP sensing probes would be stable in this matrix. Despite the excellent selectivity obtained, this sensing system can be interfered by the presence of Fe(III) ions. However, it can be greatly diminished using the CNFs as a stabilizing agent, as well as an additional dispersant. The overall results revealed that the AgNP@CNF/CNF system is a good candidate for Hg(II) detection and can be efficiently developed to paper-based platforms.
Data availability
The data that support this study are provided in the article, and the supplementary data files are available from the authors upon request.
References
Andreoli V, Sprovieri F (2017) Genetic Aspects of Susceptibility to Mercury Toxicity: An Overview. Int J Environ Res Public Health 14(1):93. https://doi.org/10.3390/ijerph14010093
Balasurya S, Syed A, Thomas AM et al (2020) Preparation of Ag-cellulose nanocomposite for the selective detection and quantification of mercury at nanomolar level and the evaluation of its photocatalytic performance. Int J Biol Macromol 164:911–919. https://doi.org/10.1016/j.ijbiomac.2020.07.119
Bhattacharjee Y, Chatterjee D, Chakraborty A (2018) Mercaptobenzoheterocyclic compounds functionalized silver nanoparticle, an ultrasensitive colorimetric probe for Hg(II) detection in water with picomolar precision: A correlation between sensitivity and binding affinity. Sens Actuators B Chem 255:210–216. https://doi.org/10.1016/j.snb.2017.08.066
Budlayan ML, Dalagan J, Lagare-Oracion JP, Patricio J, Arco S, Latayada F, Vales T, Baje B, Alguno A, Capangpangan R (2022) Detecting mercury ions in water using a low-cost colorimetric sensor derived from immobilized silver nanoparticles on a paper substrate. Environ Nanotechnol Monit Manag 18:100736. https://doi.org/10.1016/j.enmm.2022.100736
Chandraker SK, Ghosh MK, Lal M, Ghorai TK, Shukla R (2019) Colorimetric sensing of Fe3+ and Hg2+ and photocatalytic activity of green synthesized silver nanoparticles from the leaf extract of Sonchus arvensis L. New J Chem 43(46):18175–18183. https://doi.org/10.1039/C9NJ01338E
Crockett JR, Win-Piazza H, Doebler JE, Luan T, Bao Y (2021) Plasmonic detection of mercury via amalgamation on gold nanorods coated with PEG-thiol. ACS Appl Nano Mater 4(2):1654–1663. https://doi.org/10.1021/acsanm.0c03134
Dai D, Yang J, Wang Y, Yang Y-W (2021) Recent Progress in Functional Materials for Selective Detection and Removal of Mercury(II) Ions. Adv Funct Mater 31(1):2006168. https://doi.org/10.1002/adfm.202006168
Dasharathy S, Arjunan S, MaliyurBasavaraju A, Murugasen V, Ramachandran S, Keshav R, Murugan R (2022) Mutagenic, Carcinogenic, and Teratogenic Effect of Heavy Metals. Evid Based Complement Alternat Med 2022:8011953. https://doi.org/10.1155/2022/8011953
Doan V-D, Phan TL, Vasseghian Y, Evgenievna LO (2022) Efficient and fast degradation of 4-nitrophenol and detection of Fe (III) ions by Poria cocos extract stabilized silver nanoparticles. Chemosphere 286:131894. https://doi.org/10.1016/j.chemosphere.2021.131894
Driscoll CT, Mason RP, Chan HM, Jacob DJ, Pirrone N (2013) Mercury as a Global Pollutant: Sources, Pathways, and Effects. Environ Sci Technol 47(10):4967–4983. https://doi.org/10.1021/es305071v
Engström K, Ameer S, Bernaudat L, Drasch G, Baeuml J, Skerfving S, Bose-O’Reilly S, Broberg K (2013) Polymorphisms in genes encoding potential mercury transporters and urine mercury concentrations in populations exposed to mercury vapor from gold mining. Environ Health Perspect 121(1):85–91. https://doi.org/10.1289/ehp.1204951
Faghiri F, Ghorbani F (2019) Colorimetric and naked eye detection of trace Hg2+ ions in the environmental water samples based on plasmonic response of sodium alginate impregnated by silver nanoparticles. J Hazard Mater 374:329–340. https://doi.org/10.1016/j.jhazmat.2019.04.052
Foster EJ, Moon RJ, Agarwal UP, Bortner MJ, Bras J, Camarero-Espinosa S, Chan KJ, Clift MJ, Cranston ED, Eichhorn SJ (2018) Current characterization methods for cellulose nanomaterials. Chem Soc Rev 47(8):2609–2679. https://doi.org/10.1039/C6CS00895J
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21(2):885–896. https://doi.org/10.1007/s10570-013-0030-4
Gad SC, Pham T (2014) Mercury. Encyclopedia of Toxicology (Third Edition). Academic Press, P. Wexler. Oxford, pp 207–210
Gao X, Lu Y, He S, Li X, Chen W (2015) Colorimetric detection of iron ions (III) based on the highly sensitive plasmonic response of the N-acetyl-l-cysteine-stabilized silver nanoparticles. Anal Chim Acta 879:118–125. https://doi.org/10.1016/j.aca.2015.04.002
Ghosh S, Mondal A (2020) Aggregation chemistry of green silver nanoparticles for sensing of Hg2+ and Cd2+ ions. Colloids Surf a: Physicochem Eng Asp 605:125335. https://doi.org/10.1016/j.colsurfa.2020.125335
Ghosh S, Maji S, Mondal A (2018) Study of selective sensing of Hg2+ ions by green synthesized silver nanoparticles suppressing the effect of Fe3+ ions. Colloids Surf a: Physicochem Eng Asp 555:324–331. https://doi.org/10.1016/j.colsurfa.2018.07.012
Golmohammadi H, Morales-Narváez E, Naghdi T, Merkoçi A (2017) Nanocellulose in sensing and biosensing. Chem Mater 29(13):5426–5446. https://doi.org/10.1021/acs.chemmater.7b01170
Gul Z, Ullah S, Khan S, Ullah H, Khan MU, Ullah M, Ali S, Altaf AA (2022) Recent Progress in Nanoparticles Based Sensors for the Detection of Mercury (II) Ions in Environmental and Biological Samples. Crit Rev Anal Chem: 1–17. https://doi.org/10.1080/10408347.2022.2049676
Gworek B, Dmuchowski W, Baczewska-Dąbrowska AH (2020) Mercury in the terrestrial environment: a review. Environ Sci Eur 32(1):128. https://doi.org/10.1186/s12302-020-00401-x
Hashemi SA, Bahrani S, Mousavi SM, Mojoudi F, Omidifar N, Lankarani KB, Arjmand M, Ramakrishna S (2022) Development of sulfurized Polythiophene-Silver Iodide-Diethyldithiocarbamate nanoflakes toward Record-High and selective absorption and detection of mercury derivatives in aquatic substrates. Chem Eng J 440:135896. https://doi.org/10.1016/j.cej.2022.135896
Ismail M, Khan M, Akhtar K, Seo J, Khan MA, Asiri AM, Khan SB (2019) Phytosynthesis of silver nanoparticles; naked eye cellulose filter paper dual mechanism sensor for mercury ions and ammonia in aqueous solution. J Mater Sci Mater Electron 30(8):7367–7383. https://doi.org/10.1007/s10854-019-01049-x
Klemm D, Cranston ED, Fischer D, Gama M, Kedzior SA, Kralisch D, Kramer F, Kondo T, Lindström T, Nietzsche S (2018) Nanocellulose as a natural source for groundbreaking applications in materials science: Today’s state. Mater Today 21(7):720–748. https://doi.org/10.1016/j.mattod.2018.02.001
Kumar V, Mohan S, Singh DK, Verma DK, Singh VK, Hasan SH (2017) Photo-mediated optimized synthesis of silver nanoparticles for the selective detection of Iron (III), antibacterial and antioxidant activity. Mater Sci Eng C 71:1004–1019. https://doi.org/10.1016/j.msec.2016.11.013
Li WC, Tse HF (2015) Health risk and significance of mercury in the environment. Environ Sci Pollut Res 22(1):192–201. https://doi.org/10.1007/s11356-014-3544-x
Li R, Fei J, Cai Y, Li Y, Feng J, Yao J (2009) Cellulose whiskers extracted from mulberry: A novel biomass production. Carbohydr Polym 76(1):94–99. https://doi.org/10.1016/j.carbpol.2008.09.034
Lin N, Huang J, Dufresne A (2012) Preparation, properties and applications of polysaccharide nanocrystals in advanced functional nanomaterials: a review. Nanoscale 4(11):3274–3294. https://doi.org/10.1039/C2NR30260H
Lin JH, Chen SJ, Lee JE, Chu WY, Yu CJ, Chang CC, Chen CF (2022) The detection of Mercury (II) ions using fluorescent gold nanoclusters on a portable paper-based device. Chem Eng J 430:133070. https://doi.org/10.1016/j.cej.2021.133070
Logan N, Lou-Franco J, Elliott C, Cao C (2021) Catalytic gold nanostars for SERS-based detection of mercury ions (Hg2+) with inverse sensitivity. Environ Sci Nano 8(9):2718–2730. https://doi.org/10.1039/D1EN00548K
Lokanathan AR, Uddin KMA, Rojas OJ, Laine J (2014) Cellulose Nanocrystal-Mediated Synthesis of Silver Nanoparticles: Role of Sulfate Groups in Nucleation Phenomena. Biomacromol 15:373–379. https://doi.org/10.1021/bm401613h
Lokanathan AR, Uddin KMA, Chen X, Wenchao X, Xianming K, Leena SJ, Ras RHA, Rojas OJ (2015) Paper-based plasmon-enhanced protein sensing by controlled nucleation of silver nanoparticles on cellulose. Cellulose 22:4027–4034. https://doi.org/10.1007/s10570-015-0783-z
Maity M, Bera K, Pal U, Khamaru K, Maiti NC (2018) Sensing of iron (III) ion via modulation of redox potential on biliverdin protected silver nanosurface. ACS Appl Nano Mater 1(11):6099–6111. https://doi.org/10.1021/acsanm.8b01311
Marquez S, Morales-Narváez E (2019) Nanoplasmonics in paper-based analytical devices. Front Bioeng Biotechnol 7:69. https://doi.org/10.3389/fbioe.2019.00069
Morales-Narváez E, Golmohammadi H, Naghdi T, Yousefi H, Kostiv U, Horák D, Pourreza N, Merkoçi A (2015) Nanopaper as an optical sensing platform. ACS Nano 9(7):7296–7305. https://doi.org/10.1021/acsnano.5b03097
Narouei FH, Livernois L, Andreescu D, Andreescu S (2021) Highly sensitive mercury detection using electroactive gold-decorated polymer nanofibers. Sens Actuators B Chem 329:129267. https://doi.org/10.1016/j.snb.2020.129267
Ovalle-Serrano SA, Díaz-Serrano LA, Hong C, Hinestroza JP, Blanco-Tirado C, Combariza MY (2020) Synthesis of cellulose nanofiber hydrogels from fique tow and Ag nanoparticles. Cellulose 27(17):9947–9961. https://doi.org/10.1007/s10570-020-03527-6
Pomal NC, Bhatt KD, Modi KM, Desai AL, Patel NP, Kongor A, Kolivoška V (2021) Functionalized silver nanoparticles as colorimetric and fluorimetric sensor for environmentally toxic mercury ions: An overview. J Fluoresc 31(3):635–649. https://doi.org/10.1007/s10895-021-02699-z
Qing W, Zhao M, Kou C, Lu M, Wang Y (2018) Functionalization of silver nanoparticles with mPEGylated luteolin for selective visual detection of Hg2+ in water sample. RSC Adv 8(51):28843–28846. https://doi.org/10.1039/C8RA05243C
Sèbe G, Ham-Pichavant F, Ibarboure E, Koffi ALC, Tingaut P (2012) Supramolecular Structure Characterization of Cellulose II Nanowhiskers Produced by Acid Hydrolysis of Cellulose I Substrates. Biomacromol 13(2):570–578. https://doi.org/10.1021/bm201777j
Segal L, Creely JJ, Martin AE, Conrad CM (1959) An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. Text Res J 29(10):786–794. https://doi.org/10.1177/004051755902901003
Song K, Zhu X, Zhu W, Li X (2019) Preparation and characterization of cellulose nanocrystal extracted from Calotropis procera biomass. Bioresour Bioprocess 6(1):45. https://doi.org/10.1186/s40643-019-0279-z
Trang NLN, Hoang VT, Dinh NX, Tam LT, Le VP, Linh DT, Cuong DM, Khi NT, Anh NH, Nhung PT (2021) Novel Eco-Friendly Synthesis of Biosilver Nanoparticles as a Colorimetric Probe for Highly Selective Detection of Fe (III) Ions in Aqueous Solution. J Nanomater 2021. https://doi.org/10.1155/2021/5527519
Uddin KMA, Lokanathan AR, Liljeström A, Chen X, Rojas OJ, Laine J (2014) Silver nanoparticle synthesis mediated by carboxylated cellulose nanocrystals. Green Mater 2:183–192. https://doi.org/10.1680/gmat.14.00010
Vasileva P, Alexandrova T, Karadjova I (2017) Application of Starch-Stabilized Silver Nanoparticles as a Colorimetric Sensor for Mercury(II) in 0.005 mol/L Nitric Acid. J Chem 2017: 6897960. https://doi.org/10.1155/2017/6897960
Vyas G, Bhatt S, Paul P (2019) Synthesis of Calixarene-Capped Silver Nanoparticles for Colorimetric and Amperometric Detection of Mercury (HgII, Hg0). ACS Omega 4(2):3860–3870. https://doi.org/10.1021/acsomega.8b03299
Wang Y, Wang Y, Wang F, Chi H, Zhao G, Zhang Y, Li T, Wei Q (2022) Electrochemical aptasensor based on gold modified thiol graphene as sensing platform and gold-palladium modified zirconium metal-organic frameworks nanozyme as signal enhancer for ultrasensitive detection of mercury ions. J Colloid Interface Sci 606:510–517. https://doi.org/10.1016/j.jcis.2021.08.055
Yang X, Han F, Xu C, Jiang S, Huang L, Liu L, Xia Z (2017) Effects of preparation methods on the morphology and properties of nanocellulose (NC) extracted from corn husk. Ind Crops Prod 109:241–247. https://doi.org/10.1016/j.indcrop.2017.08.032
ZangenehKamali K, Pandikumar A, Jayabal S, Ramaraj R, Lim HN, Ong BH, Bien CSD, Kee YY, Huang NM (2016) Amalgamation based optical and colorimetric sensing of mercury (II) ions with silver@ graphene oxide nanocomposite materials. Microchim Acta 183(1):369–377. https://doi.org/10.1007/s00604-015-1658-6
Acknowledgements
This work was financially supported by grants from Kasetsart University Research and Development Institute—KURDI (FF(KU)9.65). We also thank the Center of Nanotechnology, Kasetsart University, and the Scientific Research Division, Faculty of Science, Kasetsart University for providing the research facilities.
Funding
This work was funded by grants from Kasetsart University Research and Development Institute—KURDI (FF(KU)9.65).
Author information
Authors and Affiliations
Contributions
JJ: Conceptualization, Investigation, Formal analysis, and Writing-original draft. PS: Material preparation and Investigation. SL: Material preparation and Investigation. BP: Material preparation and Investigation. PY: Methodology and Formal analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
The manuscript was approved by all authors for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jettanasen, J., Silakhao, P., Lohakarok, S. et al. Silver nanoparticles stabilized by nanocellulose as a sensing probe for mercury (II) detection and their response with iron (III) interference. Cellulose 31, 5609–5622 (2024). https://doi.org/10.1007/s10570-024-05963-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-024-05963-0