
Abstract
To estimate the effect of chemical treatment of wood or typify the phenotypes of genetically modified plants, it is necessary to know the coexistence mode of wood components. Herein, the average states of carbohydrate/lignin coexistence in ball-milled and cellulase-treated wood powder samples were investigated using molecular motion evaluation methods, which were originally established for phase structure analysis of simple, miscible polymer blends. Our aim was to depict the nanoscopic arrangements of the major components of wood using a unified approach. The targeted scales were 20–30 nm, which was evaluated using glass transition temperatures measured by differential scanning calorimetry, and approximately 2 nm, which was evaluated using spin–lattice relaxation times of the 1H nucleus (TH1ρ) in a rotating system measured by solid-state nuclear magnetic resonance spectroscopy. In both softwood (Japanese cypress, Chamaecyparis obtuse) and hardwood (eucalyptus, mainly Eucalyptus globulus) samples, the main components coexisted in the range 20–30 nm. The TH1ρ data revealed that the carbohydrates and lignin behaved independently in wood powders after pulverization, but their molecular motions became similar after enzymatic hydrolysis of cellulose. This meant that hemicellulose and lignin were within approximately 2 nm of each other. The results are consistent with the major findings from microscopy, thermal analysis, spectroscopy, and quartz crystal microbalance measurements. Limitations on the application of these numerical assessments were considered. We envisage that molecular motion data could be applied as an index to connect the wood composition with properties related to wood utilization.
Graphic abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The nanoscopic arrangement (Kerr and Goring 1975; Ruel et al. 1978) of hemicellulose and lignin between cellulose microfibrils (CMFs) should be directly linked to characteristics related to wood utilization, such as decay durability and efficiencies of pulping, enzymatic saccharification, and mechanical nano-fibrillation. Quantitative data for this arrangement could be used to estimate what treatments are required and improve treatment methods. Although a wide range of research has been conducted on cell wall genetics (Brown et al. 2016), a unified approach for evaluating phenotypes from the viewpoint of the nanoscopic arrangement of wood components is required.
For the nanoscopic arrangements of wood components, electron microscope images of cell and tissue structures have been reported in detail from immunolabeling (Joseleau and Ruel 1997; Awano et al. 1998, 2002; Maeda et al. 2000; Joseleau et al. 2004) and lignin chemistry (Terashima et al. 2009, 2012) experiments. Analyses of thermal transitions, including thermal softening (Irvine 1984; Kelley et al. 1987; Östberg et al. 1990; Salmén and Olsson 1998; Furuta et al. 2014) and solid-state nuclear magnetic resonance (NMR) spectroscopy (Kohn et al. 2011; Foston et al. 2012; Nishida et al. 2014), have also been carried out. Recently, adsorption of enzymes such as cellulase and hemicellulases was precisely investigated using lignocellulose nanofibers as a probe for quartz crystal microbalance analysis (Kumagai et al. 2014, 2016; Kumagai and Endo 2018). Very recently, lignocellulose nanofibers of Cryptomeria (softwood) and Eucalyptus (Eu, hardwood) were examined (Fig. 1a) (Kumagai and Endo 2018). In Cryptomeria, the CMFs were covered with glucomannan and xylan was dispersed on the surface in complexes with lignin. By contrast, in Eu, glucomannan was dotted on the CMFs and xylan was coated on the glucomannan in complexes with lignin. These findings are illustrated in Fig. 1a.

Copyright © 2018, Springer Science Business Media B.V., part of Springer Nature
Schematic drawing of microfibrils and their neighboring components in Cy and Eu samples: (a) wood powder, (b) after ball milling, and (c) after cellulase treatment. Panel (a) is reproduced with permission from a reference (Kumagai and Endo 2018).
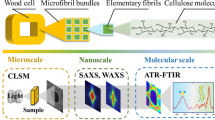
Herein, we focus on molecular motion analysis of polymers via relaxation measurements as a highly reproducible and versatile method that can provide an overview of wood composition. This method has been conveniently used for analysis of the phase structures of polymer blends. In mixtures of different polymers, cooperative movements can occur between the polymer constituents when they are in close proximity. The spatial scale of the cooperative movement varies depending on the type of molecular motion, and it is possible to evaluate the phase structure on the nanometer scale by making use of different instrumental analyses.
The glass transition behavior detected by differential scanning calorimetry (DSC) arises from cooperative movement of segments. According to a generally accepted opinion (Kaplan 1976; MacKnight et al. 1978; Olabisi et al. 1979; Utracki 1990; Nishio 2017), the level of molecular mixing to yield a single glass transition temperature (Tg) in DSC for polymer blends is supposed to be 20–30 nm as an upper limit of the possible domain size. This means that segmental motion of simple miscible blends (no cross-linking is assumed) is homogenous at this spatial scale and the different component polymer chains relax together at the Tg.
Molecular motion on the scale of several nanometers can be investigated by measuring the spin–lattice relaxation time of the 1H nucleus (TH1ρ) in a rotating system using solid-state high-resolution NMR spectroscopy (McBrierty and Douglass 1981).
By considering the presence or absence of cooperative movements and the difference in the scales evaluated in each measurement, it should be possible to numerically estimate the scale of coexistence of heterogeneous macromolecular combinations, in this case, carbohydrates and lignin. However, the methods described so far have been generally applied to simple polymer blends. For wood samples, it is necessary to consider binding between the components, which includes possible intercomponent crosslinking. Therefore, there are application limits to these numerical evaluations, and we point these out later on in the text.
In this study, to establish criteria for the nanoscopic arrangement of wood components, we aimed to numerically evaluate the average state of coexistence for carbohydrates (cellulose and hemicellulose) and lignin by molecular motion analysis via relaxation measurements. To measure the molecular motion of wood components, we removed some of the wood components and then tracked how the molecular motion changed in the remaining wood. We used powder samples of Japanese Hinoki cypress (Chamaecyparis obtuse, Cy), which is a softwood, and Eu as the raw materials. When common hydrothermal or wet chemistry methods were adopted for removing some of the wood constituents, the chemical structure of the remaining components changed (Rojo et al. 2015; Kumagai et al. 2016; Saito et al. 2016). This complicates the interpretation of the behavior of molecular motion in the original wood samples. Thus, we performed enzymatic hydrolysis, which is highly substrate specific and generally proceeds under mild conditions. Because the enzyme hydrolyzability of untreated wood powder is generally very low (Teramoto et al. 2008a, b), the samples were pretreated by ball-mill pulverization. In principle, the influence of ball milling can be considered as causing depolymerizationand destruction of higher-order structures (Endo et al. 1999; Sipponen et al. 2014). The molecular dynamics of carbohydrates and lignin in these samples were monitored by DSC and solid-state NMR spectroscopy. To date, thermal analysis, including DSC, has been mainly applied to wet wood samples (Östberg et al. 1990). In the present study, we used wood samples that were vacuum dried and conditioned in the atmosphere. Consequently, because it was not necessary to consider the influence of swelling of the components, we expected the data interpretation to be unambiguous. Estimation of the spatial scales of lignin and carbohydrates in 13C-enriched corn stover has been performed by evaluating the spin diffusion distance using solid-state NMR spectroscopy with a special pulse sequence (Foston et al. 2012). In the present study, we intended to analyze wood in as natural a form as possible. We aimed to quantitatively evaluate the coexistence of carbohydrates and lignin by investigating the influence of ball-mill pulverization and enzymatic hydrolysis on the molecular motion of each component at each stage.
Experimental
Materials
Samples of two kinds of wood were used in this research. The first was papermaking grade Eu purchased from Oji Paper Co., Ltd. (Tokyo, Japan), which was a mixture of six species with the main component being Eucalyptus globulus. The second was Cy from Maniwa city, Okayama Prefecture, Japan. The wood samples were in the form of woodchips, and a milling cutter was used to convert them to powders (particle size < 0.2 mm). The obtained powders were dried at 40 °C for 24 h in vacuo, extracted in a Soxhlet extractor with ethanol/toluene (1:2 v/v) for 24 h, and dried again at 40 °C for 24 h in vacuo. The samples were then stored at 20 °C and 20% RH in a digitally controlled desiccator [DCD-PSPS; AS ONE Corporation (Japan)]. Industrial cellulase “Meicelase” derived from the fungus Trichoderma viride was obtained from Meiji Seika Pharma Co., Ltd. (Japan) and used for enzymatic hydrolysis. Chemicals were purchased from commercial sources and used without any further purification.
Chemical composition analysis of the original wood samples
After extracting the wood powders in a Soxhlet extractor with EtOH/toluene (1:2 v/v) for 6 h to remove any extractable components, the resulting samples were analyzed to determine their holocellulose contents as the NaClO2-delignified residue (Wise et al. 1946) and Klason lignin contents as the residue insoluble in 72% sulfuric acid aqueous solution (Browning 1967). Their contents of acid soluble lignin were estimated from the absorbance at 210 nm for the soluble fraction, and their ash contents were calculated from the mass of the residue obtained after heating at 600 °C. The results for Cy and Eu are summarized in Table 1.
Ball-mill pulverization
Ball-mill pulverization was performed in a planetary ball mill (Pulverisette 6; Fritsch GmbH, Germany) equipped with a 500-mL jar loaded with 25 balls (ø 20 mm). Both the jar and balls were made of zirconia (ZrO2). A 30-g sample of the wood powder was packed in the jar. Pulverization was carried out by placing the pulverizer at room temperature (20 °C), rotating the jar at 250 rpm for 10 min, and then pausing for 10 min to prevent overheating. The total rotation time was 0, 6, 12, or 48 h. Samples were coded according to the pulverization time (e.g., Cyx for the Cy sample pulverized for x h).
Enzymatic hydrolysis
A pulverized sample with a dry mass of 3 g was added to a 200-mL eggplant flask with 80 mL of 50 mM acetate buffer. Meicelase (168 mg) was dissolved in 40 mL of 50 mM acetate buffer to give a concentration of 16 filter paper units/g of pulverized sample. This mixture was added to the suspension of pulverized wood powder. The flask containing the solid–liquid mixture was set in a water bath regulated at 45 °C and the suspension was mechanically stirred at 300 rpm for 0.5–48 h. The enzymatically hydrolyzed wood sample was washed with water, lyophilized, vacuum-dried at 40 °C for 12 h, and stored in the desiccator at 20 °C and 20% RH before use.
Constituent sugar analysis
Sugar analysis was carried out with reference to the laboratory analytical procedure published by the National Renewable Energy Laboratory (Sluiter et al. 2008). A dry sample (300 mg) and 3.0 mL of 72% sulfuric acid were placed in a 20-mL snap cup and stirred at 30 °C for 90 min. The sample was transferred to a 200-mL Erlenmeyer flask with 84 mL of Milli-Q water and autoclaved at 121 °C for 60 min. The resulting solution was filtered and the filtrate was neutralized with barium hydroxide and barium carbonate. Before analyzing the constituent monosaccharides by high-performance liquid chromatography, the neutralized hydrolyzate was passed through a membrane filter with a pore size of 0.45 µm. The constituent monosaccharides were detected by derivatization with phosphoric acid–phenylhydrazine using a pump (L-2130), reaction unit (L-5050), and fluorescence detector (L-2485) manufactured by Hitachi High-Technologies Corporation (Japan). Gradient elution for 10 µL of hydrolyzate was performed with acetonitrile/water/phosphoric acid (900/95/5 and 750/245/5 v/v/v) with a flow rate of 1.0 mL/min. The column was an Asahipak NH 2 P-50 4 E (250 × 4.6 mm, Showa Denko K.K., Japan), and the column temperature was 40 °C. The elution was performed with continuous flow to the reaction unit. The reagent passed through the reaction unit was phosphoric acid/acetic acid/phenylhydrazine (542/443/15 v/v/v) with a flow rate of 0.40 mL/min. The fluorescence detection wavelength was set to 330–470 nm.
Instrumental analysis
Elemental analysis was performed using a CHN coder JM10 (J-Science Lab Co., Ltd., Japan). Antipyrine (C11H12N2O; CAS Number, 60-80-0) was used as a standard sample.
Wide-angle X-ray diffraction (WAXD) measurements were carried out with an Ultima-IV diffractometer (Rigaku Corporation, Japan) at 20 °C in reflection mode. Nickel-filtered CuKα radiation was used at 40 kV and 30 mA. The diffraction intensity was measured between 2θ = 5–30°.
To observe the morphologies of the treated samples, a field-emission scanning electron microscope (FE-SEM) (S-4800, Hitachi High-Technologies Corporation) was used with an accelerating voltage of 1.5 kV. The samples were attached to the stage using double-sided conductive tape and then coated with osmium for 20 s at 10 mA using a Neoc-Pro osmium coater (Meiwafosis Co., Ltd., Japan) before observation.
Fourier transform infrared (FT-IR) spectra were recorded on a Spectrum 100 FT-IR spectrometer (PerkinElmer Inc., USA) with accumulation of 32 scans in the wavenumber range 400–4400 cm−1 at a resolution of 4 cm−1. A standard KBr-pellet method was used for all measurements. The sample mass fraction was 2%. The samples were dried under vacuum at 110 °C for at least 12 h before analysis.
DSC was carried out on samples (~ 5 mg) using a DSC7020 (Hitachi High-Technologies Corporation). The first heating scan was run at a rate of 20 °C/min from − 100 °C to 220 °C. After rapid cooling to − 100 °C following the first heating, a second heating scan to 250 °C was performed at 20 °C/min.
Solid-state cross polarization/magic-angle spinning (CP/MAS) 13C NMR spectroscopy experiments were conducted with a JNM-ECA500 spectrometer (JEOL Ltd., Japan) equipped with a NM-93030CPM probe (JEOL Ltd.). The measurements were recorded at 20 °C and a 13C frequency of 125 MHz. The 13C CP/MAS spectra were acquired with a spinning rate of 7 kHz, a contact time of 7 ms, a recycle delay time of 5 s, 90° pulse width of 3.3 µs, and 2048 FID signal accumulations. For quantification of the TH1ρ in the rotating frame for the wood samples, a contact time of 0.1 ms was used and the proton spin-locking time, τ, ranged from 0.1 to 8 ms. A total of 1024 scans were accumulated.
Results and discussion
In this study, we prepared Cy and Eu wood powder samples by submitting them to ball milling and enzymatic hydrolysis for different durations. We measured the sugar and elemental compositions of several samples. Then, we demonstrated that the carbohydrate/lignin (C/L) ratio for the series of samples could be conveniently estimated using FT-IR. The effect of each treatment was evaluated further using electron microscopy images and the crystallinity of the cellulose component. The molecular motions of the wood constituents were analyzed by DSC and solid-state NMR spectroscopy.
Sugar analysis
Figure 2 shows the sugar compositions of Cy12 and Eu12 before and after enzymatic hydrolysis. For the original samples (0 h), mannose and xylose were the main hemicellulose components in Cy and Eu, respectively. Over time, a remarkable decrease was observed in the glucose content in the samples, but no large decreases were observed for the contents of the other sugars. This indicates that mainly cellulose decomposition occurs with the commercially available cellulase preparation (Meicelase) used in this study and there is less digestion of hemicellulose.

Sugar compositions of Cy12 and Eu12 samples subjected to enzymatic hydrolysis for different periods. Values are given in as mass fractions (%) relative to the original wood powder samples
Elemental analysis
Elemental analysis of the wood powder samples was performed at each treatment stage (Table 2). No large changes in elemental composition were observed after pulverization. After enzymatic treatment, the carbon content increased regardless of the species, and the oxygen content decreased. These changes occurred because the oxygen-rich carbohydrate component was decomposed by the enzyme and removed, and the abundance ratio of lignin with carbon-rich phenylpropane as a constituent unit increased. By contrast, the nitrogen mass fraction did not change greatly and was consistent at ≤ 0.5% in the samples. This showed that the amount of enzyme (Meicelase) remaining in the samples was not remarkable and that the sample washing process was sufficient. Cellulase is often of concern because it can adsorb to lignin and become unproductive. However, lignin not subjected to heat treatment or chemical modification does not inhibit the enzyme function (Xu et al. 2010; Ishiguro and Endo 2015).
Estimation of the C/L ratio by FT-IR
We used FT-IR spectra to simply evaluate the degree of degradation of the carbohydrate, which was mainly cellulose according to the above sugar analysis, and the C/L ratio at each enzymatic treatment time. In both Cy and Eu, regardless of the pulverization time, the band at a wavenumber slightly higher than the 1500 cm−1 stretching vibration of the carbon–carbon bond of the aromatic rings of lignin (Pandey and Pitman 2004) became larger as the enzymatic treatment time increased (Fig. 3). By contrast, the intensity of the band at 1158 cm−1, which was attributed to the asymmetric C–O–C vibration from the carbohydrate component (Pandey and Pitman 2004), decreased as the enzymatic treatment time increased. The relative changes in the band intensities with the enzymatic treatment time reflect digestion of carbohydrate by enzymatic treatment and an increase in the relative content of lignin. We used the band intensity changes to estimate changes in the relative masses of carbohydrate and lignin in samples subjected to the enzymatic treatment for different times, with reference to the research of Pandey and Pitman (2004). Details for the procedure and validation are given in Figs. S1 and S2. Table 3 summarizes the C/L ratios for the samples. Both Cy and Eu showed an increase in the lignin content with the enzymatic treatment time.

FT-IR spectra of the wood powder samples subjected to ball milling and enzymatic hydrolysis for different periods, respectively: a Cy12, b Eu12, c Cy48, and d Eu48
Morphology
FE-SEM observation was performed to evaluate the influence of pulverization and enzymatic treatment on the morphologies of Cy and Eu. The FE-SEM images of the pulverized samples are shown in Fig. 4. In the images of the original wood powder samples, fibrous tissue structures were observed as tracheids in Cy (Fig. 4a) and wood fibers in Eu (Fig. 4b). By contrast, in the samples pulverized by ball milling, no fibrous structures were found but particles with uneven surfaces were observed (Fig. 4c, d, g, h). Among the samples, those pulverized for a long time had large particles with smooth surfaces. This is in agreement with the finding that primary particles produced at the initial stage of pulverization agglomerate with further pulverization and become secondary particles with large particle sizes (Endo et al. 1999). For the samples treated with the enzyme (Fig. 4e, f, i, j), sub-micrometer sized pores (Teramoto et al. 2008b) formed on the surface. The pores were generated by enzymatic degradation and removal of carbohydrates. The material remaining after removal of the carbohydrates appeared to include a collection of particles with sizes of several tens of nanometers. It is possible that these are spherical lignin particles polymerized in the lignification process (Terashima et al. 2009, 2012). Such particles appear to have originally filled the gaps within the three-dimensional network formed by hemicellulose and the CMFs. From these observations, we confirmed that the fibrous structure of the wood sample collapsed with ball-mill pulverization, and that enzymatic treatment resulted in formation of a structure in which carbohydrate, mainly cellulose, was removed. Schematic drawings of the microfibrils and their neighboring components for the Cy and Eu samples are given in Fig. 1. It should be noted that only removal of the polysaccharide component and changes of the dimensions of the primary particles are depicted in the figure.

FE-SEM images of wood powder samples: original a Cy and b Eu; ball-mill pulverized, c Cy12 and d Eu12; enzymatically hydrolyzed (24 h), e Cy12 and f Eu12; ball-mill pulverized, g Cy48 and h Eu48; and enzymatically hydrolyzed (24 h) i Cy48 and j Eu48
WAXD
WAXD profiles of Cy and Eu before and after ball milling are shown in Fig. S3. In the original samples, diffraction characteristic of cellulose I was mainly detected. By contrast, after ball milling for ≥ 12 h, the diffractions of cellulose I were not clear and an amorphous halo was observed. This is common behavior for CMFs on ball milling, which results in breaking of regular intermolecular hydrogen bonds, disorder of the molecular chain arrangement, deterioration of the continuity of the crystal, and conversion to a substantially amorphous solid (Endo et al. 1999). The pulverized wood powder prepared in this way consisted of amorphous carbohydrate and lignin. This change is illustrated in Fig. 1a, b. Next, we evaluated the coexistence states of the wood components.
Assignment of thermal transition behavior observed by DSC measurement
Figure 5 shows DSC thermograms in the second heating scan for Cy12, Eu12, Cy48, and Eu48. The C/L ratio was adjusted by changing the enzymatic treatment time. Thermal transition was unclear except for the exotherm from approximately 210 °C in the DSC thermograms of the Cy and Eu samples subjected to only ball-mill pulverization. A baseline shift was observed at 140–200 °C for the pulverized samples after enzymatic treatment. To elucidate the thermal events occurring here, the Cy12 enzymatically treated for 24 h was heated to temperatures where the baseline shift (180 °C) and exothermic behavior (250 °C) were detected, and the morphology was observed by FE-SEM (Fig. S4). In the SEM image of the sample heated to 180 °C, no change in morphology was seen compared with the sample heated to 150 °C. When the temperature was increased to 250 °C, irregularities on the surface of the wood powder disappeared and the surface became smooth. This morphological change was likely due to thermal denaturation, which can be interpreted as manifesting as the exothermic behavior above 210 °C in the DSC thermograms. On the other hand, glass transition occurs because of initiation of micro-Brownian motion in the amorphous part of polymer chains at temperatures above the Tg. Even if the temperature is above the Tg, the shape of the bulk sample does not change greatly as long as no external force is applied. Furthermore, the temperature region (140–200 °C) exhibiting the baseline shift was comparable that for Todo-fir milled wood lignin (midpoint Tg, 146 °C) (Uraki et al. 2012), where the thermal analysis was conducted not only by DSC but also by thermal fluidity analysis by means of thermomechanical analysis (TMA). Therefore, it is reasonable to interpret the shift of the baseline observed at around 180 °C as the glass transition. In the following, the onset Tg of each sample was estimated by DSC.

DSC thermograms of the wood powder samples with different C/L ratios in the second heating scan: a Cy12, b Eu12, c Cy48, and d Eu48
The wood components that can exhibit glass transitions are hemicellulose and lignin, and the thermal softening temperatures of hemicellulose and lignin in the dry state are assumed to be 200 °C and 150 °C, respectively (Takamura 1968). The specific heat capacity difference (ΔCp) before and after the glass transition was compared using the C/L ratio (Fig. 5). The ΔCp tended to increase as the lignin content increased. In addition, the glass transition behaviors of holocellulose (cellulose and hemicellulose), extracted lignin (prepared as described in Fig. S5), and mixtures prepared by simply mixing the powders were evaluated separately. The Tg was detected for the extracted lignin but not clearly for holocellulose, and the ΔCp increased as the proportion of lignin increased in the simple mixture (Fig. S6). Therefore, the baseline gap detected in the thermograms (Fig. 5) of the enzymatically treated wood powder samples was derived mainly from the Tg of the lignin component.
Evaluation of coexistence of the components by DSC
The onset Tg was plotted against the lignin content estimated from the C/L ratio of each sample (Fig. 6). Regardless of the wood species, the Tg shifted to a lower temperature as the lignin content increased. Such a change in the glass transition behavior accompanying a variation in the C/L ratio occurred regardless of the length of the ball-mill pulverization before the enzymatic digestion treatment. By contrast, as shown in Fig. S6, in the DSC thermograms for the mixtures of holocellulose and extracted lignin powders at different ratios, no systematic (continuous) change was observed in the temperature range of the Tg of the mixtures irrespective of the extracted lignin content. That is, when carbohydrate and lignin are merely mixed, Tg should not vary even if the lignin content is different. Therefore, the Tg drop associated with the decrease in the carbohydrate content (and increase in the lignin content) observed here means that the carbohydrates in these samples exists close to the lignin component and constrain the molecular motion of lignin.

Plots of Tg against lignin content, determined from the C/L ratio, for the treated a Cy and b Eu samples. The samples were subjected to ball-mill pulverization for 12 h or 48 h before enzymatic digestion. The results for the different lengths of ball milling are shown using solid red (12 h) and open blue (48 h) dots
As mentioned above, the scale of coexistence of the components estimated from the glass transition detected using DSC for simple miscible polymer blends is 20–30 nm (Kaplan 1976; MacKnight et al. 1978; Olabisi et al. 1979; Utracki 1990; Nishio 2017). In the present wood powder treated with pulverization and cellulase, it was necessary to consider that the carbohydrates and lignin were binding instead of simply mixing. However, it seems to be reasonable to conclude that the carbohydrates and lignin exist close to each other on a comparable scale and the carbohydrate component constrains the molecular motion of lignin.
Solid-state 13C CP/MAS NMR spectra
Solid-state 13C CP/MAS NMR spectra of Cy and Eu with different ball milling durations are shown in Fig. 7a, b, respectively. The distinct signals were assigned as follows: methyl group (18.9 ppm); C6 of carbohydrate (60.0 ppm); overlapping C2, C3, and C5 (73.2 ppm), C4 (80–90 ppm), and C1 (102.4 ppm) of the pyranose ring; and aromatic carbon (110–150 ppm) (Gil and Neto 1999; Maunu 2002). Following ball milling, the carbohydrate-derived C1 and C4 signals broadened and decreased in intensity. The C2, C3, C5, and C6 signals also broadened. The splitting of carbohydrate signals (C2, C3, C4, C5, and C6) was caused by the coexistence of crystalline and amorphous phases of cellulose, which showed that the crystal structure of the carbohydrate component disintegrated with pulverization treatment. The remarkable decrease in the C1 and C4 signals probably indicated that the β-(1 → 4) glycosidic bond in cellulose was cleaved.

Solid-state 13C CP/MAS NMR (500 MHz) spectra of the wood powder samples at 20 °C: ball-mill pulverized a Cy and b Eu; and enzymatically hydrolyzed c Cy12 and d Eu12
Figure 7c, d illustrates the 13C CP/MAS NMR spectra for Cy12 and Eu12 samples, respectively, that were enzymatically digested for different times. These results show that the C/L ratio was altered. As the enzymatic treatment time increased, the signal intensity for the carbohydrate component decreased and that of the lignin aromatic ring increased. This corresponds to the FT-IR spectroscopy results (Fig. 3), and shows that carbohydrate (mainly cellulose) was removed by enzymatic treatment and the relative lignin content increased. The remaining signal at ~ 75 ppm can be attributed to hemicellulose. This originally overlapped with the signal of cellulose.
Evaluation of the molecular motion by solid-state NMR spectroscopy
To evaluate the molecular motion of the pulverized wood samples by nuclear magnetic relaxation, the relaxation time was measured by solid-state NMR spectroscopy. In binary polymer mixtures, nuclear spin energy is exchanged between the polymers when the polymers are in close proximity, and energy averaging occurs (spin diffusion). In heterogeneous polymer mixtures where spin diffusion occurs, the relaxation times of the polymers approach, depending on the degree of homogeneity, and eventually converge. This behavior indicates that their molecular chains are moving cooperatively. Using this knowledge, the nanoscopic phase structures of polymer blend systems including cellulosics and synthetic polymers (McBrierty and Douglass 1981; Masson and Manley 1991) have been evaluated. Generally, the spin–lattice relaxation time (TH1ρ) in the rotation system of an amorphous polymer can be estimated by substituting the attenuating carbon resonance intensity into the following Debye relaxation function (single exponential function):
where M(τ) is the magnetization intensity observed at the spin locking time τ. For the signals at 56 ppm and 75 ppm derived from lignin (OCH3) and carbohydrate (C2, C3, and C5) components, respectively, if ln(M(τ)/M(0)) is plotted against τ, then TH1ρ can be obtained as the inverse of the slope (Fig. S7).
Figure 8 shows plots of TH1ρ against the ball-mill pulverization time for Cy and Eu. In both, the TH1ρ of the carbohydrate and lignin components decreased with the pulverization time; however, the degree of the decrease was significant only for the carbohydrate. Regarding the carbohydrate component, the molecular motion likely increased because of a decrease in the molecular weight and the progress of amorphization by pulverization. The relatively small change in the TH1ρ of lignin indicates that possible structural variation of its surroundings by pulverization does not remarkably affect the molecular motion of lignin at the scale (generally several nanometers) estimated by TH1ρ. Eventually, regardless of the ball-mill pulverization time, the TH1ρ of carbohydrate and lignin were not consistent in Cy and Eu. Therefore, averaging of the spin diffusion was not sufficient. That is, on the scale of several nanometers estimated by the TH1ρ measurement, the carbohydrate and lignin molecules in the pulverized wood powder samples moved independently of each other, and the components did not coexist.

TH1ρ plotted against ball milling time for Cy and Eu samples. The numerical values inserted in the figure are the L values (nm) estimated from TH1ρ when the D is set to 10−12 cm2/s in Eq. 2
If we try to interpret the data in more detail, meanwhile, the difference in TH1ρ of the lignin component of Cy before and after the pulverization was larger than that of Eu. There are two possibilities for the structural factors that led to this. One is that Cy (softwood) has a higher rate of the carbohydrate-lignin linkages which can be cleaved by pulverization than that of Eu (hardwood); In this connection, Balakshin et al. (2011) showed that in lignin–carbohydrate complex (LCC) structural units, the proportion of benzyl ether, which is prone to be cleaved by ball milling, was higher in pine (softwood) than in birch (hardwood). The other is that the lignin structure of Cy was more easily altered by pulverization than that of Eu. The current data is not sufficient to determine which of these effects is significant, and further investigation is required.
To numerically estimate the domain size of carbohydrate and lignin in the samples, an effective distance (L) over which spin diffusion can proceed in time TH1ρ was calculated. L is used to evaluate the domain sizes of polymer blends and is represented by the following equation (Clauss et al. 1993):
With the diffusion coefficient D at 10−12 cm2/s (Maunu 2002), the L values of carbohydrate and lignin components were evaluated and the numerical values are shown in Fig. 8. The L value range was 1.5–2.1 nm. That is, in the pulverized wood samples, carbohydrate and lignin moved independently of each other within these scales on average, and these components did not mix as mentioned above. Considering that the width of the glucopyranose ring is approximately 0.5 nm and the size of the benzene ring is 0.73 nm, the estimated molecular motion scales of carbohydrate and lignin roughly corresponds to about three sugar chains and two phenylpropane units, respectively. The dimension obtained here is equivalent to the spin diffusion length (~ 2 nm) estimated by solid-state 13C NMR CP SELDOM (selectively by destruction of magnetization) for 13C-enriched plants (Foston et al. 2012).
Subsequently, the effect of altering the C/L ratio by cellulase treatment on the TH1ρ was examined for Cy12 and Eu12. The TH1ρ was plotted against the lignin content obtained from the C/L ratio (Fig. 9). When the carbohydrate component was removed by a long enzymatic digestion, the TH1ρ of lignin slightly increased. Therefore, similarly to the above DSC data, the TH1ρ measurements from solid-state NMR spectroscopy indicated that the molecular motion of lignin changed on removal of carbohydrate from the wood powder samples. Considering the L value given above, this result shows that the removal of cellulose component influences the molecular motion of lignin on a scale of approximately 2 nm, because cellulose is mostly removed by the enzymatic treatment.

TH1ρ plotted against lignin contents for Cy12 and Eu12 samples subjected to enzymatic hydrolysis for different periods
After enzymatic treatment, the TH1ρ of the carbohydrate also increased, and the TH1ρ of the carbohydrate and lignin tended to approach each other more than when only ball milling was carried out. This behavior indicates that removal of the original main component, cellulose, allows the contribution of lignin molecular motion to become relatively large, and as a result, the relaxation behavior of lignin and the remaining carbohydrate (hemicellulose) becomes homogenous. The degree of homogenization was more prominent in Eu than that in Cy.
Considering the sugar analysis results (Fig. 2), after the enzymatic treatment, the hemicellulose remaining in Eu is mainly xylan, whereas that in Cy is both xylan and glucomannan. On the basis of the schematic in Fig. 1a, which shows the coexistence of hemicellulose and lignin from quartz crystal microbalance data (Kumagai and Endo 2018), it is reasonable to assume that molecular motion of lignin and xylan is homogenous in Eu. For the softwood Cy, glucomannan covers the CMFs and may exhibit molecular motion independent of lignin.
The TH1ρ results also indicate that the change in molecular motion, which could not be detected by DSC, for the carbohydrate after the enzymatic treatment could be tracked by the TH1ρ measurement.
Points to be aware of in the above discussion
We have discussed the scale of carbohydrate and lignin coexistence in wood powder samples subjected to ball-mill pulverization and enzymatic treatment. Our numerical estimates of the scale used Tg (DSC) and TH1ρ (solid-state NMR spectroscopy) evaluations of simple miscible polymer blends without crosslinking. In the wood powder samples examined here, it is high likely that there is hemicellulose-mediated binding (including possible crosslinking) between the components. Therefore, the actual coexistence scales may be larger than the estimated ones. In addition, it should be noted that the D value (10−12 cm2/s) used for determining L with TH1ρ is generally mainly applied to synthetic polymers, so there is some concern about its relevance.
However, the approach used in this study has the following features: it is not necessary to consider swelling because the samples are dry; and general purpose measurements can be performed within realistic time frames. This concept for assessing the average state of carbohydrate/lignin coexistence in wood on a unified scale should be extended to other samples in the future.
Conclusions
By evaluating Tg and TH1ρ by DSC and solid-state NMR spectroscopy, respectively, we obtained an indication of the scale of coexistence of the main component polymers of wood for powder samples of Cy and Eu subjected to ball-mill pulverization and cellulase treatment. Cellulose, hemicellulose, and lignin coexisted in the particle size range of 20–30 nm. The molecular motions of hemicellulose and lignin estimated from the TH1ρ did not become uniform by pulverization alone, but tended to converge when cellulose was removed by cellulase. This indicates that hemicellulose and lignin coexist in the range of about 2 nm. The degree of convergence appeared to be higher in Eu (hardwood) than in Cy (softwood). The validity of the interpretation of the data may be arguable because of possible binding between components. Collection of more data from other plant species and tissues would be useful. This could facilitate application of molecular motion data as an index to connect the primary molecular structure to characteristics related to wood utilization, such as decay durability and suitability for chemical pulping, mechanical nano-fibrillation, and bioethanol production. In a generalization of the evaluation of carbohydrate/lignin coexistence by this approach, to evaluate the effect of pretreatments as well as interaction type (covalent or non-covalent) would be the problem in the future.
References
Awano T, Takabe K, Fujita M (1998) Localization of glucuronoxylans in Japanese beech visualized by immunogold labelling. Protoplasma 202:213–222. https://doi.org/10.1007/BF01282549
Awano T, Takabe K, Fujita M (2002) Xylan deposition on secondary wall of Fagus crenata fiber. Protoplasma 219:106–115. https://doi.org/10.1007/s007090200011
Balakshin M, Capanema E, Gracz H et al (2011) Quantification of lignin-carbohydrate linkages with high-resolution NMR spectroscopy. Planta 233:1097–1110. https://doi.org/10.1007/s00425-011-1359-2
Brown SP, Eduardo R, Dupree R, Dupree P (2016) Folding of xylan onto cellulose fibrils in plant cell walls revealed by solid-state NMR. Nat Commun 7:13902. https://doi.org/10.1038/ncomms13902
Browning BL (1967) Methods of wood chemistry, vol 2. Wiley, New York
Clauss J, Schmidt-Rohr K, Spiess HW (1993) Determination of domain sizes in heterogeneous polymers by solid-state NMR. Acta Polym 44:1–17. https://doi.org/10.1002/actp.1993.010440101
Endo T, Kitagawa R, Hirotsu T, Hosokawa J (1999) Fine-powdering of fibrous cellulose by mechanical milling. Kobunshi Ronbunshu 56:166–173. https://doi.org/10.1295/koron.56.166
Foston M, Katahira R, Gjersing E, Davis MF, Ragauskas AJ (2012) Solid-state selective 13C excitation and spin diffusion NMR to resolve spatial dimensions in plant cell walls. J Agric Food Chem 60:1419–1427. https://doi.org/10.1021/jf204853b
Furuta Y, Okuyama T, Kojiro K, Miyoshi Y, Kiryu T (2014) Temperature dependence of the dynamic viscoelasticity of bases. J Wood Sci 60:249–254. https://doi.org/10.1007/s10086-014-1402-6
Gil AM, Neto CP (1999) Solid-State NMR studies of wood and other lignocellulosic materials. Annu Rep NMR Spectrosc 37:75–117. https://doi.org/10.1016/S0066-4103(08)60014-9
Irvine GM (1984) The glass transitions of lignin and hemicellulose and their measurement by differential thermal analysis. Tappi J 67:118–121
Ishiguro M, Endo T (2015) Effect of the addition of calcium hydroxide on the hydrothermal-mechanochemical treatment of Eucalyptus. Bioresour Technol 177:298–301. https://doi.org/10.1016/j.biortech.2014.10.135
Joseleau JP, Ruel K (1997) Study of lignification by noninvasive techniques in growing maize internodes (an investigation by fourier transform infrared cross-polarization-magic angle spinning 13C-nuclear magnetic resonance spectroscopy and immunocytochemical transmission electron microscopy. Plant Physiol 114:1123–1133. https://doi.org/10.1104/pp.114.3.1123
Joseleau J-P, Imai T, Kuroda K, Ruel K (2004) Detection in situ and characterization of lignin in the G-layer of tension wood fibres of Populus deltoides. Planta 219:338–345. https://doi.org/10.1007/s00425-004-1226-5
Kaplan DS (1976) Structure–property relationships in copolymers to composites: molecular interpretation of the glass transition phenomenon. J Appl Polym Sci 20:2615–2629. https://doi.org/10.1002/app.1976.070201001
Kelley SS, Rials TG, Glasser WG (1987) Relaxation behaviour of the amorphous components of wood. J Mater Sci 22:617–624. https://doi.org/10.1007/BF01160778
Kerr AJ, Goring DAI (1975) The ultrastructural arrangement of the wood cell wall. Cellul Chem Technol 9:563–573
Kohn B, Davis M, Maciel GE (2011) In situ study of dilute H2SO4 pretreatment of 13C-enriched poplar. Energy Fuels 25:2301–2313. https://doi.org/10.1021/ef2000213
Kumagai A, Endo T (2018) Comparison of the surface constitutions of hemicelluloses on lignocellulosic nanofibers prepared from softwood and hardwood. Cellulose 25:1–13. https://doi.org/10.1007/s10570-018-1861-9
Kumagai A, Iwamoto S, Lee S, Endo T (2014) Quartz crystal microbalance with dissipation monitoring of the enzymatic hydrolysis of steam-treated lignocellulosic nanofibrils. Cellulose 21:2433–2444. https://doi.org/10.1007/s10570-014-0312-5
Kumagai A, Lee S, Endo T (2016) Evaluation of the effect of hot-compressed water treatment on enzymatic hydrolysis of lignocellulosic nanofibrils with different lignin content using a quartz crystal microbalance. Biotechnol Bioeng 113:1441–1447. https://doi.org/10.1002/bit.25911
MacKnight WJ, Karasz FE, Fried JR (1978) Solid state transition behavior of blends. In: Paul DR, Newman S (eds) Polymer blends. Academic Press, New York, pp 184–243
Maeda Y, Awano T, Takabe K, Fujita M (2000) Immunolocalization of glucomannans in the cell wall of differentiating tracheids in Chamaecyparis obtusa. Protoplasma 213:148–156. https://doi.org/10.1007/BF01282152
Masson JF, Manley RS (1991) Cellulose/poly(4-vinylpyridine) blends. Macromolecules 24:5914–5921. https://doi.org/10.1021/ma00022a004
Maunu SL (2002) NMR studies of wood and wood products. Prog Nucl Magn Reson Spectrosc 40:151–174. https://doi.org/10.1016/S0079-6565(01)00041-3
McBrierty VJ, Douglass DC (1981) Recent advances in the NMR of solid polymers. J Polym Sci Macromol Rev 16:295–366. https://doi.org/10.1002/pol.1981.230160105
Nishida M, Tanaka T, Miki T, Shigematsu I, Kanayama K (2014) Study of nanoscale structural changes in isolated bamboo constituents using multiscale instrumental analyses. J Appl Polym Sci 131:40243. https://doi.org/10.1002/app.40243
Nishio Y (2017) General remarks on cellulosic blends and copolymers. In: Navard P (ed) Blends and graft copolymers of cellulosics: toward the design and development of advanced films and fibers. Springerbriefs in molecular science. Biobased Polymers. Springer, Cham, pp 1–16
Olabisi O, Robeson L, Shaw M (1979) Polymer–polymer miscibility. Academic Press, New York
Östberg G, Salmén L, Terlecki J (1990) Softening temperature of moist wood measured by differential scanning calorimetry. Holzforschung 44:223–225. https://doi.org/10.1515/hfsg.1990.44.3.223
Pandey KK, Pitman AJ (2004) Examination of the lignin content in a softwood and a hardwood decayed by a brown-rot fungus with the acetyl bromide method and fourier transform infrared. J Polym Sci, Part A: Polym Chem 42:2340–2346. https://doi.org/10.1002/pola.20071
Rojo E, Peresin MS, Sampson WW, Hoeger IC, Varitianen J, Rojas OJ (2015) Residual lignin on the physical, barrier, mechanical and surface properties of nanocellulose films. Green Chem 17:1853–1866. https://doi.org/10.1039/c4gc02398f
Ruel K, Barnoud F, Goring DAI (1978) Lamellation in the S2 layer of softwood tracheids as demonstrated by scanning transmission electron microscopy. Wood Sci Technol 12:287–291. https://doi.org/10.1007/BF00351930
Saito K, Horikawa Y, Sugiyama J, Watanabe T (2016) Effect of thermochemical pretreatment on lignin alteration and cell wall microstructural degradation in Eucalyptus globulus: comparison of acid, alkali, and water pretreatments. J Wood Sci 62:276–284. https://doi.org/10.1007/s10086-016-1543-x
Salmén L, Olsson AM (1998) Interaction between hemicelluloses, lignin and cellulose: structure-property relationships. J Pulp Pap Sci 24:99–103
Sipponen MH, Laakso S, Baumberger S (2014) Impact of ball milling on maize (Zea mays L.) stem structural components and on enzymatic hydrolysis of carbohydrates. Ind Crops Prod 61:130–136. https://doi.org/10.1016/j.indcrop.2014.06.052
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D, Crocker D (2008) Determination of structural carbohydrates and lignin in biomass. Laboratory Analytical Procedure (LAP). NREL/TP-510-42618
Takamura A (1968) Studies on hot pressing and drying process in the production of fibreboard. III. Softening of fibre components in hot pressing of fibre mat. Mokuzai Gakkaishi 14:75–79
Teramoto Y, Lee S-H, Endo T (2008a) Pretreatment of woody and herbaceous biomass for enzymatic saccharification using sulfuric acid-free ethanol cooking. Bioresour Technol 99:8856–8863. https://doi.org/10.1016/j.biortech.2008.04.049
Teramoto Y, Tanaka N, Lee S-H, Endo T (2008b) Pretreatment of eucalyptus wood chips for enzymatic saccharification using combined sulfuric acid-free ethanol cooking and ball milling. Biotechnol Bioeng 99:75–85
Terashima N, Kitano K, Kojima M, Yoshida M, Yamamoto H, Westermark U (2009) Nanostructural assembly of cellulose, hemicellulose, and lignin in the middle layer of secondary wall of ginkgo tracheid. J Wood Sci 55:409–416. https://doi.org/10.1007/s10086-009-1049-x
Terashima N, Yoshida M, Hafrén J, Fukushima K, Westermark U (2012) Proposed supramolecular structure of lignin in softwood tracheid compound middle lamella regions. Holzforschung 66:907–915. https://doi.org/10.1515/hf-2012-0021
Uraki Y, Sugiyama Y, Koda K et al (2012) Thermal mobility of β-O-4-type artificial lignin. Biomacromol 13:867–872. https://doi.org/10.1021/bm201772v
Utracki L (1990) Polymer alloys and blends: thermodynamics and rheology. Hanser, Munich/New York
Wise LE, Murphy M, D’Addieco AA (1946) Chlorite holocellulose, its fractionation and bearing on summative wood analysis and on studies on the hemicelluloses. Pap Trade J 122:35–43
Xu J, Cheng JJ, Sharma-Shivappa RR, Burns JC (2010) Lime pretreatment of switchgrass at mild temperatures for ethanol production. Bioresour Technol 101:2900–2903. https://doi.org/10.1016/j.biortech.2009.12.015
Acknowledgments
We are grateful to Prof. Takahisa Nishizu of Gifu University for his help with sugar analysis. Elemental analyses, SEM observations, and IR and NMR spectroscopy were performed using the facilities at the Division of Instrumental Analysis, Life Science Research Center, Gifu University. This work was financially supported by Grant-in-Aids for Challenging Exploratory Research (Grant No. 16K14955 to Y.T.) and Scientific Research (A) (Grant No. 17H01480 to Y.T.) from the Japan Society for the Promotion of Science and JST-Mirai Program (Grant No. JPMJMI18E3). We thank Gabrielle David, Ph.D., from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Katsu, N., Endo, T. & Teramoto, Y. Evaluation of the average state of carbohydrate/lignin coexistence in wood by analysis of molecular motion. Cellulose 27, 41–56 (2020). https://doi.org/10.1007/s10570-019-02792-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-019-02792-4