
Abstract
Solvents that produce cellulose nanocrystals (CNCs) and promote cellulose fibrillation are of current interest. In this work, CNCs were fabricated from cotton at 80 and 100 °C using deep eutectic solvents (DESs) having choline chloride/oxalic acid dihydrate (OA) ratios of 1:1, 1:2 and 1:3. To investigate the side effects of the fabrication, the crystal structure and morphology of micro-sized treated cellulose together with nano-sized CNCs were analyzed by X-ray diffraction, field emission scanning electron microscopy and atomic force microscopy. OA promoted the formation of carboxyl groups on the C6 positions of molecules on the hydrophilic (1–10) lattice planes, causing extensive fibrillation of cellulose and disruption of surface layers on (110) and (200) planes. Lower crystallinity and lamellar structures for CNCs with mild treatment were observed after mechanical disintegration and subsequent lyophilization, which was ascribed to van der Waals forces and hydrogen bonding between adjacent crystalline cellulose chains, accelerating the self-assembly into cellulose macrofibrils. This work is discussed in light of cellulose supramolecular structures that are modified from CNC fabrication via DES treatment, with a view to enhancing the efficacy of treatment by understanding the variations that arise in cellulose structure from a green solvent.
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Among various lignocellulosic resources, cotton fiber has the highest content of cellulose as much as 95% (Kettering and Conrad 1942; Reddy and Yang 2009). Cotton cellulose is utilized in the textile industry extensively where its fiber length is advantageous. Cotton has a highly crystalline cellulose component in contrast with wood cellulose (Saito et al. 2006). Thus, it has long been considered as a model for crystalline studies of cellulose Iβ, a stable form with a two-chain unit cell having monoclinic symmetry (Sugiyama et al. 1990).
Cellulose nanoparticles have received increasing attention due to their high strength and stiffness combined with low weight, biodegradability and renewability (Klemm et al. 2011). Cellulose nanocrystals (CNCs) are rod-like or whisker shaped particles, often prepared from micro-fibrillated cellulose through a variety of chemical treatments. CNCs have a high aspect ratio (50–500 nm length, 3–5 nm width) and crystal structure that varies in different materials. Cotton CNCs generally have longer length and higher aspect ratio than wood CNCs, perhaps due to the longer fibers of the original cellulose. A typical CNC is composed of the cellulose Iβ allomorph with a hexagonal cross-section with multiple surfaces (Moon et al. 2011; Ding and Himmel 2006). The conventional methods for preparing CNCs include acid hydrolysis, enzymatic hydrolysis and some oxidation reactions that can cause hydrolysis of the amorphous regions in cellulose chains. The treatments are often followed by high pressure mechanical or ultra-sonic disintegration (Nechyporchuk et al. 2016; Ling et al. 2018). However, these methods still have some problems such as energy and time consumption and use of toxic chemicals. Thus, in recent years, reports on the preparation of cellulose nanoparticles utilizing deep eutectic solvents (DES), a new kind of solvent consisting of hydrogen bond acceptors (HBA) and hydrogen bond donors (HBD), are of much interest. can be synthesized using either anionic, cationic, or nonionic species (Chen et al. 2016; Tang et al. 2017). Moreover, DES can be prepared with 100% atom-economy, less toxicity and lower energy-consumption than ILs.
Sirviö et al. (2015) made the first cellulose nanoparticles by DES treatment. Choline chloride-urea DES with a molar ratio of 1:2 was used as a non-hydrolytic pretreatment of birch cellulose at 100 °C which was followed by micro fluidizer disintegration. The lengths of the cellulose nanofibrils (CNFs) ranged from 15 to 200 nm with widths of 2–5 nm. The crystallinity after mechanical nanofibrillation showed some reduction even though there was no obvious polymorphic change from cellulose Iβ. Similarly, urea-LiCl DES was used as a reaction medium in the succinylation of wood cellulose (Selkälä et al. 2016).
CNCs from wood cellulose were obtained using choline chloride (ChCl) and oxalic acid dihydrate (OA) (Sirviö et al. 2016). The width and length of the crystals were adjusted by changing the reaction conditions like molar ratio of DES, reaction time and temperature. The crystallinities of CNCs were generally higher than CNFs made from DES treatment, thought to be a result of the dissolution of more amorphous regions. The resulting nano-fibrillated cellulose had a high carboxyl content (0.20–0.28 mmol/g). CNCs synthesized with ChCl and OA at molar ratio of 1:1 were used as an oil in water emulsions stabilizer (Laitinen et al. 2017). They had higher carboxyl content and lower conductivity compared to CNCs prepared by acid hydrolysis, while the crystallinity and crystal sizes of the CNCs were similar. A mild microwave DES treatment to produce CNCs from cotton cellulose was reported recently (Liu et al. 2017). Their CNCs were made with ChCl and OA including a 3 min microwave-assisted treatment. This approach produced CNCs that had higher Segal crystallinity (82%) and better thermal stability compared to CNCs fabricated by acid hydrolysis. The intra-molecular hydrogen bond interactions in cotton cellulose are thought to be disrupted by competing hydrogen bond formation between chloride ions and the hydroxyl groups in cellulose. The OA modified the surface of the CNCs, increasing the proportion of carboxyl moieties, and promoted the dispersion of the CNC suspensions.
The ChCl and OA DES system has been reported to produce CNCs successfully from different sources. However, the effects of treatment conditions, such as temperature and DES ratio of HBD/HBA on the properties of CNCs have been examined only to a limited extent, and studies are limited on resulting supramolecular modifications of cellulose during the DES treatment. In this work, CNCs were fabricated from cotton cellulose with ChCl and OA and subsequently processed by a high-power ultra-sonication method. The structural variations of DES treated cellulose were detected on both micro and nano scales providing further insight on the mechanisms of CNC production by ChCl/OA DES treatments.
Experimental section
Materials
American Upland raw cotton fiber (Gossypium hirsutum L.) was acquired from the national registry. After a typical bleaching procedure (Hebeish et al. 2009), it was ground with a Wiley mill (Thomas-Wiley Mill Co., Philadelphia, PA, USA) passing through a 60 mesh screen. ChCl and acetone were purchased from Sigma Aldrich (St. Louis, MS, USA) and OA was purchased from JT Baker (Philipsburg, NJ, USA). All chemicals used in this study were either analytical or reagent grade.
Deep eutectic solvent treatment and fibrillation of cotton cellulose
ChCl and OA were mixed with molar ratios of 1:1, 1:2 and 1:3 to make DESs. The DESs with different molar ratios were synthesized at 80 °C in a round bottom flask for 1 h until a clear viscous liquid was obtained. Then, 1 g of bleached cotton cellulose (C0) was added into 100 mL of the clear liquid with magnetic stirring for 2 h. The treatment temperatures were 80 °C and 100 °C. After treatment, the mixture was filtered and washed by acetone and deionized water until the sample was at neutral pH.
The treated cellulose were mixed in deionized water followed by a high-power ultrasonic homogenizer (MSK-USP-3 N) with a maximum of 300 W, 20 kHz. The suspensions were centrifuged at 5000 rpm for 15 min and adjusted to a concentration of 0.05%. The nanocrystals from DES treatment at eighty and one hundred degrees with ChCl:OA ratios of 1:1, 1:2 and 1:3 were correspondingly denoted by NDE1, NDE2, NDE3, NDH1, NDH2 and NDH3.
Attenuated total reflectance–Fourier transform infrared (ATR–FTIR) spectroscopy
Chemical transformations of cotton cellulose treated at different conditions were characterized by ATR–FTIR spectroscopy (Bruker Optics, Billerica, MA, USA). The diameter of the attached zinc selenide ATR crystal was 15 mm. The samples were oven dried and scanned over the spectral range of 600–3600 cm−1 with 32 scans per spectrum.
Optical transmittance, particle size and zeta potential
The optical transmittance values of the CNC suspensions were collected at wavelengths from 300 to 750 nm at a scan speed of 200 nm/min using a UV–vis spectrophotometer (Shimadzu UV-2600). Particle size distribution and Zeta potential were determined by a Nano-ZS90 zeta-sizer (Malvern Instruments Ltd., UK) equipped with 4 mW He–Ne laser (633 nm) and 90° collecting optics. Each sample was measured in triplicate and analyzed using Malvern Dispersion Technology Software 4.20.
Morphology
AFM analyses for CNC suspensions of 0.05% concentration were conducted using an Agilent AFM series 5500 (Agilent Technologies) instrument. The sample (5 μL) was deposited on a freshly cleaved mica surface and dried for 10 min. AFM images were observed at resonance frequencies ranging from 150 to 240 kHz with further analysis by Gwyddion software (2.50 version). Treated cellulose and freeze-dried CNCs were also observed with a FEI Quanta 3D FEG FE-SEM. The accelerating potential was 5 kV, with a beam current of 20 mA. The samples were sputter coated with a thin 3 nm layer of gold–palladium using a Leika EM ACE600.
X-ray diffraction
XRD measurements were performed at room temperature with a PANalytical Empyrean diffractometer using Cu Kα-radiation (1.5418 Å) and a PIXcel3D 1 × 1 detector. X-ray patterns were acquired for cotton cellulose, treated cellulose and CNCs lyophilized by a Freezemobile 25XL freeze dryer (VirTis, Gardiner, NY, and USA) for 48 h. The patterns were further normalized and then analyzed using the pseudo-Voigt peak shape with the MAUD Rietveld program (Materials Analysis Using Diffraction, version 2.7). For crystalline and amorphous phases, the cellulose Iβ crystal information file and cellulose II crystal information file (by changing to small crystal to simulate the amorphous content of cellulose) were employed to fit the experimental patterns (Nishiyama et al. 2002; French 2014). The crystallinity was calculated from the area of the calculated pattern for crystalline cellulose divided by the sum of the areas for crystalline and amorphous regions. The d-spacings were calculated from refined unit cell dimensions and crystallite sizes perpendicular to different lattice planes were calculated using the Scherer Eq. (1).
where λ is the X-ray wavelength; Bhkl is the angular full-width at half maximum intensity (FWHM) in radians of the (hkl) line profile; θ is the scattering angle (Holzwarth and Gibson 2011).
Results and discussion
The fabrication of CNCs and initial characterization
Table 1 lists the ChCl/OA ratios and conditions of the DES treatment. A simple heating method was used, in contrast with a previously reported microwave heating approach (Liu et al. 2017). However, the yields of treated cellulose under different molar ratio and temperature conditions were 80–90%. The 10–20% loss of cotton cellulose is ascribed to the dissolution of amorphous parts of cellulose in the DES under increased temperature. The ratio of ChCl and OA had no obvious effect on the yield whereas the treated cellulose at 100 °C yielded lower amounts compared to treated cellulose treated at 80 °C. It is suggested that increased temperature accelerates the breakdown of hydrogen bonding and the hydrolysis of amorphous cellulose.
Subsequent to preparative ChCl/OA treatment of the cotton fibers, treated cellulose was subjected to conversion to CNCs by sonication. The CNCs from treatments at different temperatures and ratios resulted in a series of aqueous suspensions as shown in Fig. 1a. Determinations of the average particle length and charge of the CNCs are portrayed in Fig. 1b and listed in Table 2. Assessment of the average particle length of CNC suspensions at a dilute concentration (0.05%) revealed that the average length of CNCs was reduced by the presence of more OA during the DES treatment (Table 2). Higher temperature may also contribute to CNCs having a smaller distribution range as well as smaller average sizes (Fig. 1b). The results agree with previous conclusions that harsher treatment conditions tend to yield shorter CNCs (Martínez-Sanz et al. 2011). In terms of aspect ratio measured by dividing the length by the height of each sample, the results herein reported were slightly higher than cotton CNCs derived from sulfuric acid hydrolysis (Morais et al. 2013). This result may be ascribed to the fibrillation procedure for DES treatment in contrast with acid hydrolysis. Thus the particles retain a lower average height than expected for CNCs. Despite the long crystals of NDE1, the aspect ratio increased when higher OA ratio and temperature were applied. A similar trend reported earlier revealed a high aspect ratio and lower particle size for CNCs from DES treatment at 120 °C (Sirviö et al. 2016). Thus, DES fibrillation of cellulose microfibrils (CMF) enables qualitative improvements in CNCs.

a The CNCs (concentration: 0.05%) from DES and high-power ultra-sonic treatment with different ChCl/OA molar ratio and temperature. b Average length distribution of CNCs. Higher molar ratio and temperature tend to result in longer nanocrystals in dispersion
UV–vis light transmittance spectra showed the dispersion of CNCs in suspensions (Fig. S1). At similar concentrations following mechanical disintegration, the differences in transmittance depended on the particle size and dispersion of CNCs. The high transmittance of CNCs showed well-dispersed particles in the suspension after the DES treatment followed by high-power ultra-sonication (Han et al. 2013). Comparing different suspensions, CNCs treated with highest DES molar ratio (ChCl:OA = 1:3) and temperature had increased light transmittance at lower wavelength regions which may be explained by smaller particle sizes and fewer microfibril fragments for NDE3 and NDH3. Notably, increasing temperature during the treatment may help to prevent the aggregation of CNCs, retaining small particle sizes.
The morphological information about CNC suspensions was obtained from AFM images (Fig. 2). All the samples displayed thin needle-like nanoparticles even though there was some aggregations for samples NDE1 and NDH1 (Fig. 2a and d). Aggregation was caused by the drying process after drop casting which can form a CNC network (Fang et al. 2016). NDE3 and NDH3 images both showed better dispersion than the other samples regardless of treatment temperature (Fig. 2c, f). The dispersion of NDE3 and NDH3 samples confirmed the SEM results that higher acid ratios in DES facilitate the fibrillation of cellulose and dissolution of amorphous regions. The average heights of CNCs were measured by scanning the white bar areas in topography images that were representative for single nanocrystals (Fig. S2). As seen in Table 2, the heights of CNCs (the range of 4–10 nm) decreased with the increasing DES molar ratio.

AFM topography images of CNCs from different DES treatments: NDE1 (a), NDE2 (b), NDE3 (c), NDH1 (d), NDH2 (e), and NDH3 (f). Height profiles were determined along white bars in each image (see Fig. S2)
Zeta potential can be used to measure the electrostatic repulsive forces, which affect the stability of CNC suspensions. Zeta potential values for the most of the CNCs ranged from − 31 to 36 mV. Thus, most of the samples are highly negatively charged due to the high surface charge density of carboxyl groups on the CNC surfaces (Table 2). However, the lowest zeta value appeared for NDH1 (− 19.77 mV), may be explained based on the lowest aspect ratio (Table 2) and a smaller surface area as identified in the AFM images (Fig. 2). This higher zeta potential for NDE3 and NDH3 indicates that increased proportions of OA in DES systems are essential for promoting the stability of CNC suspensions especially during a high temperature treatment.
Chemical characterization of DES treated cellulose by ATR–FTIR
The differences in dispersion properties of CNCs may be attributed to different temperature and ChCl/OA molar ratios before mechanical disintegration. The chemical changes of DES treated cellulose under treatment with different conditions were analyzed by ATR–FTIR (Fig. 3). Treated cellulose showed peaks similar to C0 at 2900 cm−1 and 895 cm−1 attributed to C–H stretching and β-glycosidic linkages of cellulose (Lee et al. 2016), They indicate typical structures of cellulose Iβ that were treated by DES with different conditions (Table 1). The peak assigned to carbonyl stretch of carboxylic acid group at 1736 cm−1 appeared after the DES treatment, and this is consistent with the increasing band intensity at 1429 cm−1 due to the –COO− symmetric stretching vibration (Cuba-Chiem et al. 2008; Rosa et al. 2010). The increased intensity of the bands assigned to carboxyl groups correlates with higher OA ratio and treatment temperatures. However, the increase in carboxylation is limited to extended treatment at higher temperature (treatment of more than 100 °C and 2 h). This observation is consistent with previous work that noted over-esterification and formation of diesters (Sirviö et al. 2016). Also notable is a study on cellulose hydrolysis by molten OA, which showed a higher degree of substitution and carboxyl content within 60 min of the reaction (Li et al. 2017).

ATR–FTIR analysis of Wiley-milled bleached cotton cellulose (C0) and cellulose treated by ChCl/OA DES with different ratio and temperature: a wavelength range of 600–3600 cm−1, b zoomed in range of 1250–1800 cm−1, bands at 1429 cm−1 and 1736 cm−1 indicate the formation of carboxylic groups
The varying amount of carboxyl group content was further confirmed by the increased band intensity at 1153, 1119, 1061 and 1028 cm−1 respectively assigned to –CO stretching, C5–C6 stretching, –COC bending and C6–O bending vibrations (Zhbankov et al. 2002; Ilharco et al. 1997; Cael et al. 1974). The higher content of carboxyl groups principally formed at the C6 position in the treated cellulose are thought to promote the subsequent fibrillation and disintegration of cellulose (Saito et al. 2006; Isogai et al. 2011).
Morphological observation of treated cellulose with FE-SEM
The morphology of treated cellulose fibers was investigated by FE-SEM. The Wiley-milled sample (C0) micrograph displayed intact and straight fiber (Fig. S3). The microfibrils were ordered and embedded in the fibers. After DES treatment, it is apparent that increased temperature and higher ratios of OA gave rise to coarser structures on the treated cellulose fiber surfaces (Fig. 4A). This is evident as seen in contrasting the effect on the cotton fiber surface at treatment conditions of ChCl/OA, 1:2 at 80 °C in contrast with ChCl/OA 1:3. Cross sectional scission of the treated cellulose fibers under the same ratios (1:2 vs. 1:3) suggested separation of the microfibrillar networks as is evident in Fig. 4A(c). This observation is consistent with the DES and heat treatment swelling the CMF and disrupting hydrogen bonding within the inter-molecular cellulose chains. The cross sectional images of fibers (at higher magnification) taken from a higher temperature treatment showed single microfibrils [Fig. 4A(f)]. The surfaces of fibers as viewed in the longitudinal direction undergo shrinkage as evident from contraction of surface topography and exposure of randomly arranged microfibrils, which becomes more pronounced with increased temperature treatments (Fig. 4B). As discussed earlier by the growing bands at 1429 cm−1 and 1736 cm−1 from FTIR results, higher OA ratios result in an increase of carboxyl groups on the cellulose chains which was possibly caused by the esterification of hydroxyl groups of cellulose with OA. It could take place at the surface of cotton fibers and subsequently enable the accelerated separation and exposure of CMF. Thus, enhanced hydrolysis of intramolecular linkages is enabled, which prepares the fibrous network for mechanical formation of CNCs.

A FE-SEM images of the ends of treated cellulose fibers: DE1 (a), DE2 (b), DE3 (c), DH1 (d), DH2 (e) and DH3 (f). Coarser surfaces and individualization of the microfibrils are observed for higher molar ratio and temperature treatment. B FE-SEM observations of longitudinal direction of treated cellulose fibers: DE1 (a), DE2 (b), DE3 (c), DH1 (d), DH2 (e) and DH3 (f). Circles locate exposed microfibrils and arrows point to the shrinkage and wrinkles
FE-SEM observation of freeze-dried CNCs
After freeze drying, SEM images showed the morphology of CNCs prepared under different DES treatments (Fig. 5). NDE1 and NDH1 presented lamellar structures composed of aligned thin membranous layers with widths of 5–10 μm. The layered structure appears smooth on the surface and in parallel alignment, which may be explained by the van der Waals forces and hydrogen bonding between adjacent crystalline cellulose chains (Han et al. 2013; Salminen et al. 2017; Deng et al. 2009). It has previously been shown that connected cellulose chains self-assemble into larger cellulose macro fibrils and further self-organized in parallel to form a film-like structure during freeze drying. Thus, an explanation of the structural arrangement observed may be given based on alignment of ice crystals with cellulose nanocrystals compacted in the interstitial spaces of crystalline ice sheets. The growth of ice crystals as the gradient decrease of temperature created a lamellar microstructure oriented in parallel. As the sublimation process proceeds, cellulose nanocrystals align in parallel and are concentrated in the space between the ice crystals resulting in a shape consistent with the the ice morphology.

FE-SEM images of freeze-dried CNCs: NDE1 (a), NDE2 (b), NDE3 (c), NDH1 (d), NDH2 (e) and NDH3 (f). The CNCs NDE1 and NDE2 tend to aggregate to smooth lamellar structures
CNCs treated with higher OA ratios exhibited layer exfoliation and porous structure indicating that they were not assembled as well by the lyophilization as NDE1 and NDH1 (Fig. 5b, c, e, f). Considering the different ranges of particle sizes for each sample, it was hypothesized that the gaps between CNCs of NDE3/NDH3 were not small enough for tight hydrogen bonding interactions. As a result, the appearance of disorganized exfoliation or incompletely formed layers. Moreover, the relatively higher mutual electrostatic repulsion revealed by Zeta potential analysis prevented the self-assembly of CNCs during the lyophilization (Deville et al. 2006).
Crystal structure analyses
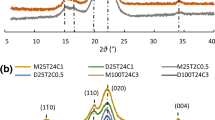
The X-ray diffraction patterns of C0, treated cellulose and CNCs are presented in Fig. 6. Typical diffraction peaks emerged at approximately 2θ = 14.8°, 16.6° and 22.6° for (1–10), (110) and (200) lattice planes of cellulose I, as well as a small peak at around 2θ = 34.5° assigned to the (004) plane. The patterns suggested that both DES treatment and following mechanical ultra-sonication kept the cellulose I allomorph of original cotton cellulose. A small shoulder at 2θ = 20.5° for C0 and the CNCs was attributed to the superimposed reflections of (012) and (102) lattice planes (Fig. 6a). The shoulder peaks seemed to disappear for high OA ratio treated samples (DE3, DH3, NDE3, and NDH3) indicating the more randomly arranged cellulose crystals instead of the preferred orientation in the samples treated with milder conditions (French 2014). The crystallinity values are presented in Fig. 7a. Sample C0 exhibited high crystallinity (93.37%) mainly owing to the originally high amount of crystalline cellulose for raw cotton materials and the bleaching procedure which extracted the wax and dissolved some amorphous parts of cellulose (Nam et al. 2016). Treated cellulose showed high crystallinity similar to C0 except for some decline for DE3 (88.16%) and DH3 (85.87%). Thus, DES treatment retained both crystalline and amorphous parts of samples as well as the high yield discussed previously. The cellulose chains with carboxyl groups on the C6 positions had a limited effect on the crystalline proportion of treated cellulose even though there was some decrease with higher acid concentration. The crystallinity values of CNCs were respectively 75.27%, 73.85%, 70.81%, 74.98%, 75.29% and 67.29%, representing substantial reduction compared to C0 and treated cellulose. The values were similar to results obtained under different reaction time on wood pulp, but lower than microwave/DES treated cotton CNC calculated by Segal method (Sirviö et al. 2016; Liu et al. 2017). The process of mechanical disintegration of cellulose in water was believed to decrease crystallinity due to the strong shear forces peeling the cellulose chains to some extent. Meanwhile, the subsequent freeze drying lowered the numbers of intermolecular hydrogen bonds between crystalline cellulose molecular chains, resulting in the decreased crystallinity of CNCs (Isogai 2013; Iwamoto et al. 2007).

XRD patterns of a C0 and treated cellulose and b freeze-dried CNCs

Crystalline properties of C0, treated cellulose and CNCs: cellulose crystallinity (a), relationship between d-spacings and crystallite sizes of lattice planes of (1–10) (b), (110) (c) and (200) (d)
To further elucidate the crystal structure variations during the DES treatment, crystallite sizes and d-spacings for the main lattice planes of cotton cellulose were plotted (Fig. 7). After DES treatment, changes were not clear for the crystallite size perpendicular to the (1–10) lattice plane for the treated cellulose samples, while the crystallite sizes were visibly reduced perpendicular to the (110) and (200) planes. The thermal environment during the treatment was responsible for the reduction, as well as the expansion of the d-spacings for the (200) plane (Kim et al. 2001; Hori and Wada 2005). Thus, it can be concluded that there is some loss of crystalline layers (about 0.4 nm per layer) for treated cellulose. Interestingly, the expansion of d-spacings on (110) and (200) planes was more visible for samples treated at 80 °C rather than 100 °C, indicating that higher acid concentration of DES played the role of impeding crystalline expansion. The high ratio of OA agglomerating on the surface of crystalline cellulose may cause a steric hindrance effect and thus prevent the growth perpendicular to hydrophobic (200) plane (Kuribayashi et al. 2016).
After mechanical fibrillation and freeze drying, CNCs displayed distinctive trends different from the treated cellulose. Crystallite size decreased for the (1–10) plane especially for NDH2 (5.528 nm) and NDH3 (5.523 nm). It took place accompanied with the reduction of d-spacings, suggesting that the lyophilization may lead to the compression of cellulose crystallites in this direction. There was some growth for the crystallite size perpendicular to the (110) lattice plane as well as the (200) plane that was more noteworthy. For example, it is known that the high-power mechanical fibrillation would disturb the crystalline arrangement of cellulose chains, shrinking the apparent crystallite sizes. However, the prominent size increase perpendicular to the (200) reflection together with lower d-spacings of CNCs proved co-crystallization during freeze drying. It has been reported that the more hydrophobic (200) surfaces have a stronger tendency to aggregate when subjected to drying (Idström et al. 2013). The co-crystallized layers may be derived from the fractions of crystallites in the solution during DES treatment or mechanical disintegration. The high values of NDE1 (5.384 nm) and NDH1 (5.397 nm) indicated that the crystallization was favored when there were fewer carboxyl groups in CNCs. It also supported the SEM observation that NDE1 and NDH1 tended to form flat lamellar structures.
Models on the production of CNCs by DES treatment and mechanical fibrillation were present in Fig. 8. In the treatment system, OA connected well with the (1–10) plane of treated cellulose which maintained ordered packing of cellulose chains. The high hydrophilicity of this reflection satisfied the linkage of OA, forming a large amount of carboxyl groups. The gathering of OA around the hydrophobic (200) plane and (110) which is less hydrophilic than (1–10) plane caused the disruption of hydrogen bonding of glucose residues on the surface (Matthews et al. 2006). Thus, the surface layer may be loosened which is supported by the decreased crystallite sizes. Mechanical disintegration is the critical step for making CNCs. But subsequent lyophilization is expected to have significant effects on the crystal structure. With the presence of carboxyl groups, the crystallite size shrank perpendicular to the (1–10) plane is mainly attributed to the decreased d-spacing. The fractions of glucose units tended to recrystallize, remarkably enlarging the crystallite sizes perpendicular to the (200) lattice plane direction. As a result, the ideal shapes of CNCs were preserved despite some interference on surface layers due to the effects of shearing force, moisture content and temperature.

Proposed schematic of cellulose crystalline variations after ChCl/OA DES treatment and subsequent mechanical fibrillation into CNCs. The arrows refer to the changing directions of crystallite sizes after freeze drying
Conclusions
In the present work, cotton cellulose was treated with ChCl/OA DES system followed by high-power ultra-sonication. The OA group plays an important role in esterification of hydroxyl groups in C6 positions on the hydrophilic (1–10) lattice plane, thus breaking the hydrogen bonding between microfibrils and making more fibrillation. The breakdown of hydrogen bonding was more significant with higher acid ratio (ChCl: OA = 1:3) in DES treatment, which may produce CNCs with better dispersion and higher aspect ratio. Mechanical disintegration and freeze drying resulted in less crystallinity than C0 and treated cellulose as well as the sheet-like layer structure for CNCs with fewer carboxyl groups. Increasing crystallite sizes suggested the recrystallization from cellulose fractions and adjacent layers on the surface of (200) lattice planes. The detection of crystalline variations of cotton cellulose could be a beneficial way for comprehending the mechanism of CNC production by the environmentally friendly DES treatment through the whole procedure.
References
Cael JJ, Koenig JL, Blackwell J (1974) Infrared and raman spectroscopy of carbohydrates: part IV. identification of configuration-and conformation-sensitive modes for d-glucose by normal coordinate analysis. Carbohydr Res 32:79–91
Chen L, Zhu JY, Baez C, Kitin P, Elder T (2016) Highly thermal-stable and functional cellulose nanocrystals and nanofibrils produced using fully recyclable organic acids. Green Chem 18:3835–3843. https://doi.org/10.1039/C6GC00687F
Cuba-Chiem LT, Huynh L, Ralston J, Beattie DA (2008) In situ particle film ATR FTIR spectroscopy of carboxymethyl cellulose adsorption on talc: binding mechanism, pH effects, and adsorption kinetics. Langmuir 24:8036–8044
Deng M, Zhou Q, Du A, van Kasteren J, Wang Y (2009) Preparation of nanoporous cellulose foams from cellulose-ionic liquid solutions. Mater Lett 63:1851–1854
Deville S, Saiz E, Nalla RK, Tomsia AP (2006) Freezing as a path to build complex composites. Science 311:515–518
Ding S-Y, Himmel ME (2006) The maize primary cell wall microfibril: a new model derived from direct visualization. J Agric Food Chem 54:597–606. https://doi.org/10.1021/jf051851z
Fang W, Arola S, Malho JM, Kontturi E, Linder MB, Laaksonen P (2016) Noncovalent dispersion and functionalization of cellulose nanocrystals with proteins and polysaccharides. Biomacromolecules 17:1458–1465. https://doi.org/10.1021/acs.biomac.6b00067
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896. https://doi.org/10.1007/s10570-013-0030-4
Han J, Zhou C, Wu Y, Liu F, Wu Q (2013) Self-assembling behavior of cellulose nanoparticles during freeze-drying: effect of suspension concentration, particle size, crystal structure, and surface charge. Biomacromolecules 14:1529–1540
Hebeish A, Hashem M, Shaker N, Ramadan M, El-Sadek B, Hady MA (2009) New development for combined bioscouring and bleaching of cotton-based fabrics. Carbohydr Polym 78:961–972
Holzwarth U, Gibson N (2011) The Scherrer equation versus the ‘Debye-Scherrer equation’. Nat Nanotechnol 6:534
Hori R, Wada M (2005) The thermal expansion of wood cellulose crystals. Cellulose 12:479
Idström A, Brelid H, Nydén M, Nordstierna L (2013) CP/MAS 13C NMR study of pulp hornification using nanocrystalline cellulose as a model system. Carbohydr Polym 92:881–884. https://doi.org/10.1016/j.carbpol.2012.09.097
Ilharco LM, Garcia AR, Lopes da Silva J, Vieira Ferreira LF (1997) Infrared approach to the study of adsorption on cellulose: influence of cellulose crystallinity on the adsorption of benzophenone. Langmuir 13:4126–4132
Isogai A (2013) Wood nanocelluloses: fundamentals and applications as new bio-based nanomaterials. J wood Sci 59:449–459
Isogai A, Saito T, Fukuzumi H (2011) TEMPO-oxidized cellulose nanofibers. Nanoscale 3:71–85
Iwamoto S, Nakagaito AN, Yano H (2007) Nano-fibrillation of pulp fibers for the processing of transparent nanocomposites. Appl Phys A 89:461–466
Kettering J, Conrad C (1942) Quantitative determination of cellulose in raw cotton fiber. Simple and rapid semimicro method. Ind Eng Chem Anal Ed 14:432–434
Kim D-Y, Nishiyama Y, Wada M, Kuga S, Okano T (2001) Thermal decomposition of cellulose crystallites in wood. Holzforschung 55:521–524
Klemm D, Kramer F, Moritz S, Lindström T, Ankerfors M, Gray D, Dorris A (2011) Nanocelluloses: a new family of nature-based materials. Angew Chem Int Ed 50:5438–5466
Kuribayashi T, Ogawa Y, Rochas C, Matsumoto Y, Heux L, Nishiyama Y (2016) Hydrothermal transformation of wood cellulose crystals into pseudo-orthorhombic structure by cocrystallization. ACS Macro Lett. pp 730–734. https://doi.org/10.1021/acsmacrolett.6b00273
Laitinen O, Ojala J, Sirviö JA, Liimatainen H (2017) Sustainable stabilization of oil in water emulsions by cellulose nanocrystals synthesized from deep eutectic solvents. Cellulose 24:1679–1689. https://doi.org/10.1007/s10570-017-1226-9
Lee CM, Chen X, Weiss PA, Jensen L, Kim SH (2016) Quantum mechanical calculations of vibrational sum-frequency-generation (SFG) spectra of cellulose: dependence of the CH and OH peak intensity on the polarity of cellulose chains within the SFG coherence domain. J Phys Chem Lett 8:55–60
Li D, Henschen J, Ek M (2017) Esterification and hydrolysis of cellulose using oxalic acid dihydrate in a solvent-free reaction suitable for preparation of surface-functionalised cellulose nanocrystals with high yield. Green Chem 19:5564–5567. https://doi.org/10.1039/c7gc02489d
Ling Z, Zhang X, Yang G, Takabe K, Xu F (2018) Nanocrystals of cellulose allomorphs have different adsorption of cellulase and subsequent degradation. Ind Crops Prod 112:541–549
Liu Y, Chen W, Xia Q, Guo B, Wang Q, Liu S, Liu Y, Li J, Yu H (2017) Efficient cleavage of lignin-carbohydrate complexes and ultrafast extraction of lignin oligomers from wood biomass by microwave-assisted treatment with deep eutectic solvent. Chemsuschem 10:1692–1700. https://doi.org/10.1002/cssc.201601795
Martínez-Sanz M, Lopez-Rubio A, Lagaron JM (2011) Optimization of the nanofabrication by acid hydrolysis of bacterial cellulose nanowhiskers. Carbohydr Polym 85:228–236
Matthews JF, Skopec CE, Mason PE, Zuccato P, Torget RW, Sugiyama J, Himmel ME, Brady JW (2006) Computer simulation studies of microcrystalline cellulose Iβ. Carbohydr Res 341:138–152
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev. 40:3941–3994
Morais JPS, de Freitas Rosa M, Nascimento LD, do Nascimento DM, Cassales AR (2013) Extraction and characterization of nanocellulose structures from raw cotton linter. Carbohydr Polym 91:229–235
Nam S, French AD, Condon BD, Concha M (2016) Segal crystallinity index revisited by the simulation of X-ray diffraction patterns of cotton cellulose Iβ and cellulose II. Carbohydr Polym 135:1–9. https://doi.org/10.1016/j.carbpol.2015.08.035
Nechyporchuk O, Belgacem MN, Bras J (2016) Production of cellulose nanofibrils: a review of recent advances. Ind Crops Prod 93:2–25. https://doi.org/10.1016/j.indcrop.2016.02.016
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082
Reddy N, Yang Y (2009) Properties and potential applications of natural cellulose fibers from the bark of cotton stalks. Bioresour Technol 100:3563–3569
Rosa MF, Medeiros ES, Malmonge JA, Gregorski KS, Wood DF, Mattoso LHC, Glenn G, Orts WJ, Imam SH (2010) Cellulose nanowhiskers from coconut husk fibers: effect of preparation conditions on their thermal and morphological behavior. Carbohydr Polym 81:83–92
Saito T, Nishiyama Y, Putaux J-L, Vignon M, Isogai A (2006) Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 7:1687–1691
Salminen R, Baccile N, Reza M, Kontturi E (2017) Surface-induced frustration in solid state polymorphic transition of native cellulose nanocrystals. Biomacromolecules. https://doi.org/10.1021/acs.biomac.7b00463
Selkälä T, Sirviö JA, Lorite GS, Liimatainen H (2016) Anionically stabilized cellulose nanofibrils through succinylation pretreatment in urea–lithium chloride deep eutectic solvent. ChemSusChem. pp 3074–3083. https://doi.org/10.1002/cssc.201600903
Sirviö JA, Visanko M, Liimatainen H (2015) Deep eutectic solvent system based on choline chloride-urea as a pre-treatment for nanofibrillation of wood cellulose. Green Chem 17:3401–3406. https://doi.org/10.1039/C5GC00398A
Sirviö JA, Visanko M, Liimatainen H (2016) Acidic deep eutectic solvents as hydrolytic media for cellulose nanocrystal production. Biomacromolecules 17:3025–3032. https://doi.org/10.1021/acs.biomac.6b00910
Sugiyama J, Okano T, Yamamoto H, Horii F (1990) Transformation of Valonia cellulose crystals by an alkaline hydrothermal treatment. Macromolecules 23:3196–3198
Tang X, Zuo M, Li Z, Liu H, Xiong C, Zeng X, Sun Y, Hu L, Liu S, Lei T, Lin L (2017) Green processing of lignocellulosic biomass and its derivatives in deep eutectic solvents. Chemsuschem 10:2695. https://doi.org/10.1002/cssc.201701012
Zhbankov RG, Firsov SP, Buslov DK, Nikonenko NA, Marchewka MK, Ratajczak H (2002) Structural physico-chemistry of cellulose macromolecules. Vibrational spectra and structure of cellulose. J Mol Struct 614:117–125
Acknowledgments
The authors gratefully acknowledge financial support by National Key Research and Development Program of China (2017YFD0601004) and Chinese Scholarship Council (CSC No. 201706510045). We also appreciate Dr. Mohammad Saghayezhian of LSU Shared Instrument Facility for technical assistance with the XRD experiments.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ling, Z., Edwards, J., Guo, Z. et al. Structural variations of cotton cellulose nanocrystals from deep eutectic solvent treatment: micro and nano scale. Cellulose 26, 861–876 (2019). https://doi.org/10.1007/s10570-018-2092-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-2092-9