
Abstract
Cellulose, being an abundantly available natural biopolymer, has a high potential to replace synthetic fibre. However, a paucity of knowledge and awareness on the use of cellulose, and agricultural waste base in particular, hampers efforts to harness this highly biodegradable resource to its full potential. This study aimed to extract and characterise cellulose obtained from pineapple (Ananas comosus) leaves of cultivar Josapine. Chemical composition analysis found that the pineapple leaves contain 11.8% lignin and 55.1% holocellulose, of which 32.6% is alpha cellulose. This extracted cellulose is light yellow to pure white in colour. The extraction of cellulose trace amounts of lignin was found to be between 0.4 and 2.8%. SEM micrograph of pineapple leaves shows fibrils 36 µm in diameter while the sample cellulose has defibrillated fibres with an average diameter ranging from 4.28 to 3.12 μm. Viscosity tests show the molecular weight (MW) at 3.8671 × 10−4 to 1.0560 × 10−4 and the degree of polymerisation of 447–107, respectively. Characterisation using X-ray diffraction (XRD), Raman and Fourier-transform infrared (FTIR) spectroscopy shows the cellulose samples are cellulose I. As compared to control, XRD data shows considerably higher percentage of crystallinity (82.90–85.41%) in all bleached sample with crystal size ranging from 3.564 to 3.807 nm. In conclusion, the extraction of cellulose from pineapple leaves using 2% NaOH and hypochlorite bleaching retains the original structure of cellulose in the pineapple leaves. The unique properties of cellulose microfibre from Josapine pineapple leaf fibres have great potential for further utilisation in various industries.
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, research focusing on the use of more sustainable and environmentally friendly biomaterial has grown rapidly. There is enormous interest in utilising agricultural waste residues as the main source of cellulose, the world’s most abundant and renewable natural crystalline biopolymer (Aziz et al. 2016; Zugenmaier 2001). Lignocellulosic biomass such as coconut husk fibres, sugarcane bagasse, banana rachis, mulberry bark, soybean pods, rice straw, soy hulls, and cornstalks have been studied as resources in the production of cellulose micro/nano fibres (CMNF) (Akhtar et al. 2015). The promising performance of CMNF and their abundance has encouraged many pieces of research to produce a variety of high-value products within different industries, e.g. in the food industry as stabiliser, dietary fibre, fat replacer and texturing agent (Buffiere et al. 2017; Serpa et al. 2016; Shi et al. 2014); in cosmetics as a thickener and coating agent (Ludwicka et al. 2017); in the medical and pharmaceutical as wound dressing, prosthesis, blood filter, drug excipient and purging agent (Ludwicka et al. 2017; Tang et al. 2017); in construction as mouldable, lightweight, high-strength composites (Mohammadkazemi et al. 2015); and in manufacturing of high-quality paper products (Li and Lee 2017), vehicles (Sanoj and Balasubramanian 2014) and industrial paint coatings (Khelifa et al. 2016) as well as alternative materials for electronics (Sabo et al. 2016).
Cellulose is a linear natural homopolymer composed of a stereo-regular chain of d-anhydroglucose units linked by 1, 4, β-glycosidic bonds at C1 and C4 carbon atoms. This hydrophilic polymer presents with three hydroxyl groups [primary hydroxymethyl (CH2OH) at the C-6 position and secondary OH at the C-2 and C-3] of different reactivities (Fan et al. 1987; Khalil et al. 2012). These hydroxyl groups play an important role in placing the crystal packing and aligning the chains parallel to each other. In its native form, cellulose is characteristically called cellulose-1 which is divided into two distinct crystalline phases or allomorphs, namely cellulose Iα and Iβ (Atalla and Vanderhart 1984). The ratio of these allomorphs is found to vary from one plant species to another. The crystallographic structure of these allomorphs has been determined with great accuracy due to profuse hydrogen bonding system (Nishiyama et al. 2002). This intramolecular and intermolecular network create a stable complex structural hierarchy of cellulose with a high axial stiffness (Moon et al. 2011).
CMNF can be obtained through mechanical treatments such as high-intensity ultrasonication (Chen et al. 2011; Wang and Cheng 2009), high pressure homogenisation (Li et al. 2012), microfluidisation (Wang et al. 2015), cryocrushing (Chakraborty et al. 2005), grinding (Chen et al. 2017) and/or chemical treatments such as alkali treatment/bleaching (Ishak et al. 2016), peroxide (Owolabi et al. 2017), enzymatic hydrolysis (Chen et al. 2017), acetylation (Werner et al. 2017), mercerisation (Sharma et al. 2015), and graft copolymerisation (Lizundia et al. 2016). Typically, pre-treatments such as alkali treatment and bleaching are used, mainly to remove large amounts of lignin, hemicellulose, wax and oils which cover the external surface of the fibre cell wall (Ishak et al. 2016). Alkali treatment defibrillates the external cellulose microfibrils, exposes short-length crystallites and depolymerises the native cellulose structure. Subsequently, bleaching leads to oxidisation and solubilisation of chromographoric groups of the lignin, the natural dyes in the fibre (Abraham et al. 2011). The time of reaction during the chemical treatment is also plays an important role in the resulting morphology and properties of CMNF (Klemm et al. 2011; Rosa et al. 2010).
It is known that the source variation is clearly illustrated in terms of crystallite size, microfibril angle, amount of non-cellulosic residues and quality (Deepa et al. 2015; Klemm et al. 2011). Therefore, the isolation of CMNF from different lignocellulosic biomass sources in different geological regions is relevant in order to efficiently compare and exploit these resources (Deepa et al. 2015). Pineapple leaf fibre (PALF) is a lignocellulose residue produced in biomass every year by tropical fruit companies, though currently only very small portions are being used in energy production and for feedstock (Cherian et al. 2010). Due to its high lignin and cellulose content, PALF is difficult to degrade, which contributes to an unfavourable environmental impact (Cherian et al. 2011). About 70–85% of PALF is glucose polymer cellulose, much of which is in crystalline structure. Another 6–19% is hemicellulose, an amorphous polymer usually composed of galactose, arabinose, xylose, and mannose. The remainder is mostly lignin (4–15%), wax (< 4%), ash (1–5%), and lesser amounts of mineral. Normally, it contains 11–15% moisture (Asim et al. 2015). With high α-cellulose content and a comparatively low microfibrillar angle (14°), it forms microfibrils with high tensile strength (Cherian et al. 2011). The cellulosic plant fibre, which is surrounded by a matrix of lignin and hemicelluloses, exists in an amorphous state and with the association of crystalline phases through both intermolecular and intramolecular hydrogen bonding, which explains its high thermal degradation. Partly as a result of such bonds, CMNF from PALF has superior mechanical properties and is deemed as an excellent potential reinforcement in composite matrices (Neto et al. 2015; Ya’acob and Aziz 2017).
Due to its sweet taste, marketability, and relatively high market price, pineapple from Josapine cultivar in Malaysia peat soil has emerged as one of the best pineapple varieties (Shamsudin et al. 2007). Therefore, there is a need to carry out an in-depth study of the chemical and morphological characteristics of Malaysian Josapine PALF and evaluate in detail the structural and molecular properties of these fibres. Through these characterisations, it will be possible to make a precise assessment of the market for different applications, as mentioned above. Attempts have been made in the current work to optimise the bleaching time for cellulose extraction after the alkali treatment. The morphology and physicochemical properties, which include the crystallinity and molecular structure of bleached cellulose microfibrils, are characterised to unravel the underlying chemical alteration after the bleaching treatment. Additionally, a viscosity test is conducted to determine the MW and the degree of polymerisation of the CMF.
Experiments
Materials
The pineapple leaf fibres (PALF) used in the present study were obtained from a local plantation in Pekan Nanas, Pontian, Johor, Malaysia. The plant species used was Ananas cosomus (belonging to the Bromeliaceae family) from the Josapine variety (Fig. 1). The leaves were sorted, cleaned and cut into small pieces (3–5 cm). The cut pineapple leaves were oven-dried for 24 h at 80 °C. The dried leaves were then grounded and sieved through a 1 mm mesh in a Wiley mill (Model 4, Thomas Wiley laboratory mill, Philadelphia, PA, USA) for about 10-15 min.

Photographs of the Malaysian Josapine pineapple plant
Other reagents used were: sulphuric acid (H2SO4, 95.0–98.0% w/v), sodium acetate, sodium hydroxide (NaOH), sodium chlorite (NaClO2, technical grade, 80%), glacial acetic acid and potassium bromide (KBr). All of these were supplied by Sigma–Aldrich (MO, USA). The chemicals used were reagent grade and were used as received.
Chemical composition of the cellulose fibre
The lignin content of the PALF was determined using the conventional Klasson’s TAPPI T222 om-6 method (Tappi 2002) and the holocellulose analysis was performed according to Wises’s sodium chlorite method (Wise et al. 1946), with some modifications adapted from (Ibrahim et al. 2014). The content of α-cellulose was measured based on the amount of insoluble holocellulose residue in a 17.5% w/v NaOH solution at room temperature whereas the hemicellulose content was calculated by subtracting the α-cellulose part from the holocellulose content.
Isolation of CMF cellulose microfibre (CMF)
The method of extraction of cellulose fibre performed in this present study involved multistep procedures which included alkaline treatment followed by bleaching in accordance with procedures suggested by Abraham et al. (2011) and Cherian et al. (2011), with some modifications. The bleaching solution was prepared by mixing sodium hypochlorite (NaClO, 4% w/v) with the mixture of acetic acid and NaOH (2% w/v). Briefly, the holocellulose obtained was immersed in bleaching solution at the ratio of 20:1. The base mixture was heated at 85 ± 5 °C with different retention times (1, 2, 3 and 4 h), as shown in Table 1. The insoluble residue (cellulose) was then collected and rinsed with distilled water until the filtrate was neutral (pH 7). The cellulose fragment was then oven-dried at 80 °C for 24 h.
Determination of Cellulose Molecule Weight and the Degree of Polymerisation
The cellulose product was diluted in cadoxen suspension at different viscosities (3.0 × 10−3, 2.5 × 10−3, 1.5 × 10−3 and 1.0 × 10−3 g mL−1). The test was performed using Ubbelodhe viscometer tube capillary (Lauda, Germany). The degree of polymerisation (DP) of all the samples was determined by plotting the cellulose viscosity versus Huggins and Kreamer equations (Eqs. 1 and 2) as follows:
where [η], ηsp, and ηrel are intrinsic, specific and relative viscosity, respectively. k′H and k′K are Huggins’s and Kraemer’s constants, respectively, whereas c is the cellulose viscosity (Alves et al. 2010; Cai and Zhang 2005). The interception point between the equations is used to calculate the MW using Mark-Houwink’s equation (Eq. 3):
whereas the polymerisation degree (DP) of the PALF sample is determined using Eq. 4:
Characterisation methods
Particle size measurement
The particle size and size distribution of the PALF samples were determined using a Microtrac particle size analyser (Model HRA9320-X100, Nikkiso Co. Ltd., Tokyo, Japan). The fibre (0.5 g) was dispersed in ethanol in the sample holder and measured using the Static Light Scattering method. The average particle size was expressed as the mean diameter ± standard deviation (SD) of three measurements. Span index, a measurement to test the polydispersity of the beads, was calculated using Eq. 5:
where D0.1 is the diameter of the bead calculated at the 10th percentile of undersized particles, D0.5 is the diameter of the bead calculated at the 50th percentile of undersized particles, and D0.9 is the diameter of the bead calculated at the 90th percentile of undersized particles.
Microscopic analyses using Scanning electron microscopy (SEM)
The morphology of the PALF samples was analysed with a Scanning Electron Microscope (Hitachi S-3400 N Variable Pressure SEM) without gold sputtering.
X-ray diffraction (XRD)
X-ray equatorial diffraction profiles of the PALF and CMFs were collected using X-ray Powder Diffraction from a Bruker Model D8 Advance Diffractometer (Bruker AXS GmbH, Karlsruhe, Germany), with CuKα radiation (λ = 0.1541 nm) at the operating voltage and current of 30 kV and 20 mA, respectively. The diffraction intensities recorded were between 2 and 80° (2θ angle range). The crystalline index was calculated using Segal’s method (Eq. 6):
where It is the total intensity of the 200 peak for cellulose 1β (at about 2θ = 22°-23°) and Ia is the amorphous intensity between the 110 and the 200 at 2θ 18° (French and Santiago Cintrón 2013; Nam et al. 2016). The average size of crystallite was calculated from the Scherrer equation (Eq. 7):
where L is the size of crystallite, perpendicular to the plane, λ is X-ray wavelength, H is the full width half maximum (FWHM, in radians) and θ is the corresponding Bragg angle (Jin and Pascal Kamdem 2009).
Fourier transform infrared (FTIR) spectroscopy
The cellulosic samples were finely ground into powder by a fibre microtome and then blended with KBr followed by pressing the mixture into ultra-thin pellets. The FTIR spectra of all samples were recorded using Perkin-Elmer Spectrum 400, (Norwalk, CT, USA) in the range of 650–4500 cm−1 with a resolution of 4 cm−1.
Raman spectroscopy
Raman spectra were acquired using a Renishaw inVia Raman microscope with a 785 nm laser and 10 s integration time at 100% of laser power on the sample (approximately 93 mW). All samples were prepared by compressing the fibres into cellulosic pellets by means of hydraulic pressure.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 6.01 (GraphPad Software Incorporated, USA). For CMF characterisation, One-way Analyses of Variance (ANOVA) were used to determine significant differences amongst the samples. Differences between means were detected using Bonferroni’s post hoc test. For all analyses, differences were considered as significant at p < 0.05.
Results and discussion
Extraction yield and chemical composotion of the CMF
The chemical extraction process is known to affect the chemical content of the PALF. The initial stage of extraction in alkaline condition hydrolyses the raw fibres to remove the hemicellulose, minerals, silica and ash (Deepa et al. 2015). The later stage of bleaching aims to remove the remaining lignin where the reaction occurs at low pH and high temperature (Acharjee et al. 2017). Figures 2a, 1b–d show the physical aspects of the non-treated PALF and treated PALF (after subsequent bleaching for up to 4 h). Visually, the change in colour (to white) was obviously noted in bleached cellulose indicating the removal of most of the initial non-cellulosic components. The extraction yield and chemical composition of the fibre from PALF are shown in Table 1. The extraction yield of cellulose sample based on dry mass: mass ratio with the raw sample is between 70.9 and 73.5%. There are gradual reductions in the dry mass of the sample during the soaking period of up to 4 h due to removal of impurities. The present study, however, had to continue up to 4 h of bleaching time due to the significant drop (p > 0.05) to 65.2% (data is not shown) in the extraction yield after 5 h. The bleaching period probably had resulted the unfavourable degradation of cellulose. PALF consist of 32.6% cellulose, 55.1% holocellulose (of which 22.5% are hemicellulose) and 11.8% lignin. The contents of α-cellulose were increased by up to 52.93% whilst the hemicellulose and lignin were reduced by 22.2% and 11.4%, respectively. For treated PALF, the contents of lignin after 4 h of bleaching were substantially reduced by 2.4% when compared to 1 h bleaching time. The findings demonstrate that the subsequent alkali and bleaching process further reduced the percentage of lignin and increased the percentage of α-cellulose component present in the CMF, whereas the hemicellulose content was not affected by this treatment. The lignin content were deteriorate due to the cleavage of the ester-linked substances of lignin which were extensively removed (Deepa et al. 2015). It can also be concluded that the lignin removal rate was directly related to bleaching retention time. However, there is a large variation in the value of cellulose content of PALF from the existing literature. Some differences can be attributed to sample preparation and extraction method, as well as the geographic and climatic conditions (Chandrasekhar et al. 2003). However, there is already an extensive literature, both technical and descriptive, on the negative impact of bleaching on the cellulose fibre (Stephen and Parungao 2003). dos Santos et al. (2013) reported there will be up to 18% loss in cellulose content during the purification process that may be attributed to cellulose breakdown during bleaching and loss of material during mass transfer and filtering process. These authors also found 74.5% cellulose, 20.4% hemicellulose and 8.72% lignin in the extracted Brazillian PALF using the same chemical method. Meanwhile, (Cherian et al. 2010) found PALF to have a high percentage of cellulose content (98.63%) with 0.8% lignin as it undergoes steam explosion and bleaching. Since there are non-selective type bleaching agents, the alteration of cellulose and degradation by excessive or inadequate bleaching invariably lowers its strength and resistance to chemicals, and thus its value, in view of its inferior physical properties (Stephen and Parungao 2003).

Photograph of a ground untreated pineapple leaves (RAW), and treated pineapple leaves with b 1, c 2 and d 4 h of bleaching
Morphological structure of CMF
SEM observation of the raw PALF and the treated PALF was used to learn about the size and surface features of the cellulose fibres. Based on Fig. 2a, the sample of raw leaf displayed a group of gummed fibril-shape fibres with an average diameter of 36.00 µm indicating that the fibre bundles from Josaphine cultivar are amongst the finest as their diameter is less than 100 µm (Mohamed et al. 2010). The surface of the raw cellulose was found to be considerably submerged with amorphous cellulose and other impurities. The fibre surface is rough and irregular, composed of waxes and other encrusting substances including hemicelluloses, lignin and cellulose. However, the alkaline and bleaching treatments led to obvious differences in the morphology (Fig. 2b–e) with no significant differences observed in CMF samples over varying bleaching time. The NaOH treatment of cellulose also resulted in the formation of micro-sized cellulose fibrillations. Plus, the CMF displayed a neat and smoothened surface after 4 h of bleaching, corresponding to the removal of impurities and amorphous cellulose. This resulted in the formation of gaps between the PALF, splitting the fibres into smaller sizes.
The bleaching solution containing 2% w/v NaOH was in good agreement with the study conducted by Owolabi et al. (2018) who demonstrated the effects of combined alkali-bleaching treatment on the fibrils microstructure of screw-pine leaves. Bleaching the alkali-treated leaf with 2 wt% NaOH for 120 min is effective in removing the hemicellulose and lignin content, resulting in fibril with a smoother surface. While a lower concentration of NaOH may not be very effective in removing the hemicellulose and lignin content, the bleaching treatment at high concentration of NaOH causes rapidly oxidized lignin, which results in degradation of the lignin content. The SEM micrograph of the bleached leaf at 6 and 8 wt% NaOH for 120 min revealed that although all hemicellulose and lignin were completely removed, the rectangular structure of the fibre became wider, was no longer intact, and was separated from its bundle, indicating signs of degradation.
Microfibre size distributions have been found to be an important determinant of the isolation method. Generally, a smaller-sized sample provides better extraction of cellulose and removal of other components. Table 2 illustrates the distribution of bead size (μm) and span index (polydispersity or width of size distribution) of the four CMF samples with different bleaching times (S1, S2, S3 and S4) following alkaline treatment. Despite the varying bleaching times, the sizes of cellulose fibre, ranging between 3.12 and 4.28 μm, were not found to be significantly different (p > 0.05). The microfibrils have approximately the particle sizes of 10 μm length and 2 μm diameter with an aspect ratio of ~ 5 (Messiry 2014). A further isolation procedure from previous studies confirmed it can be used to gradually disintegrate the micro-sized cellulose fibres into nano-fibres (Cherian et al. 2010). Therefore, the term cellulose micro/nanofibrils (CMF) is used in this study. With regards to the span index, there was also no significant difference (p > 0.05) between the samples. The corresponding values were higher than 1, indicating non-uniformity of fibril size, but the decreasing number showed the impact of bleaching towards uniform size distribution after the treatment.
Cellulose polymerisation study
During alkaline treatment and bleaching, the removal of hemicellulose and lignin may also impact the cellulose chain. This will cause variations in MW and polymerisation degree of the cellulose sample. Based on the graph plotted in Fig. 3 and data from Table 2, there is a correlation between treatment time, MW and degree of polymerisation of the CMF. Tbe lowest degree of polymerisation (107) and MW of 1.05 × 103 was assessed for samples after 4 h of bleaching. It should be noticed that in this case, the lower values of the degree of polymerisation were associated with longer bleaching times as compared to the other samples with shorter bleaching times.

Scanning electron microscopy (SEM) micrograph of cellulose microfibres from a pineapple leaf fibre (RAW) and treated cellulose fibres after b 1, c 2, d 3 and e 4 h of bleaching
Relative crystallinity of the CMF
XRD studies of the raw and treated PALF were carried out to investigate the crystalline behaviour of the CMF. The relative crystallinity and XRD diffractogram of the raw and treated PALF are shown in Fig. 4. The samples exhibited three diffraction peaks of Bragg’s angle (2θ) at ranges of 14.7° to 14.9°, 16.5° to 16.8° and 22.5° to 23.0° for planes 1–10, 110 and 200 respectively. This corresponded to the characteristic diffraction peaks of the cellulose I as described by French (2014). Accordingly, the values for native cellulose were 14.8°, 16.7° and 23.0° for planes 1–10, 110 and 200, respectively. As the bleaching time increased, the peaks were presented at approximately the same 2θ value while no peak were observed for cellulose II characteristic peak (1–10) at around 12.2° (French 2014). This indicated that the extensive bleaching up to 4 h and alkali treatment with 2% w/v NaOH, did not contributed to cellulose II transformation. Higher NaOH concentration is needed to disrupt the cellulose lattice with high lateral order mode to trigger such conversion (Yue et al. 2013).

Plot of intrinsic viscosity (η) against cellulose concentration (c) of treated cellulose fibres (S1, S2, S3 and S4)
The Crystallinity Index (CI) may vary depending on the source of cellulose and extraction method (Park et al. 2010; Wada et al. 1997). Using Segal’s method showed that the CI of bleached cellulose (up to 85.41%) was higher than the raw cellulose (62.82%) in a time-dependent manner, as shown in Table 3. The increase in crystallinity after subsequent alkali and bleaching treatment has been reported by several authors. Azraaie et al. (2016) demonstrated that the crystallinity of the cellulose isolated from banana pseudostem increases during the chemical treatment and the most highest was found when 2% (v/v) of NaOH is used. High crystallinity can be achieved by removal of hemicellulose and lignin, which existed in the amorphous regions, leading to the realignment of cellulose molecules (Mtibe et al. 2015; Rambabu et al. 2016).
The crystallite size of cellulose samples was calculated using the Scherrer equation. The crystallite size of bleached CMF was larger with a diameter range of 3.564–3.807 nm compared to the crystallite size of raw leaf fibre (1.582 nm). To explore the crystallite size of the extracted fibre in its nanoscale size through microscopy examination (such as Environmental SEM, Atomic Force Microscopy (AFM) and Transmission Electron Microscopy), a further isolation procedure (e.g. acid hydrolysis, steam explosion, etc.) is required to gradually disintegrate the micro-sized cellulose fibres into nano-fibres. Our XRD analysis on calculated crystallite size is in accordance to an AFM examination study conducted by dos Santos et al. (2013) on a PALF sample subjected to acid hydrolysis for 30 and 60 min, which exhibited the crystal diameter of 4.45 and 4.18 nm, respectively. The XRD diffraction profiles of all samples obtained were in accordance with the cellulose 1β characteristic from the International Centre for Diffraction Data (ICDD #00-056-1718) (Fawcett et al. 2008). With reference to a report by Wada et al. (1997), monoclinic structure of 1β is dominantly abundant in the plant. The z-function value of less than 0, obtained from a distance of two adjacent planes (d), demonstrated a dominant monoclinic structure (1β) in all bleached samples (Table 4).
Structural arrangement by FTIR-Raman analysis
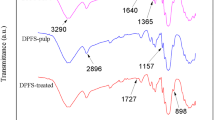
CMF samples were analysed using FTIR and Raman spectroscopy to observe the changes in the chemical composition of fibres before and after chemical treatments, as shown in Fig. 5. For the CMF sample, the strong and broad absorption band represented by a peak at 3423 cm−1 was assigned to a vibration-stretching hydrogen bond of O–H groups at C-6 followed by a strong asymmetric absorption of O–H groups at C-2. The presence of a peak at 3352 cm−1 represented vibration stretching of O–H groups at C-3, forming intramolecular hydrogen bonds with oxygen (O-5) in a chain. An intermolecular hydrogen bond between two adjacent glucose chains gave rise to another vibration-stretching peak of O–H groups between C-6 and C-3 at band 3301 cm−1. This characteristic peak was largely determined by inter- and intra-molecular bonding with hydroxyl groups (OH) (Kondo 1997). Alkali treatment reduced hydrogen bonding due to the removal of the hydroxyl groups in reaction with sodium hydroxide (Oh et al. 2005). This resulted in an increase of the –OH concentration, evident from the increased intensity of the peak between 3300 and 3500 cm−1 bands compared to the untreated fibre (Łojewska et al. 2005).

Diffractogram of cellulose microfibres from raw pineapple leaf fibre (RAW) and treated cellulose fibres (S1, S2, S3 and S4)
Furthermore, the peak at 2897 cm−1 was associated with the symmetrical and unsymmetrical stretching band presented in treated PALF whereas unsymmetrical stretching peak at band 2851 cm−1 of methyl group (-CH) was only observed in raw PALF, which was associated with the presence of lignocellulose. The peak of the latter gradually disappeared over bleaching time. The depression of absorption peaks at 1731, 1516 and 1237 cm−1 in CMF was attributed to the carbonyl group (from hemicellulose and lignin), aromatic –C=C– (from lignin) and –COO of acetyl group (from hemicellulose), respectively. With reference to Field Emission Scanning Electron Microscopy (FESEM) images, it is worth pointing out that the structure of the raw samples might have been altered because of the re-motion of amorphous cellulose and other impurities after bleaching. Due to the high cellulose–water interaction, despite the careful drying process, another peak, assigned for water, occurred at 1640 cm−1 in all cellulose samples. Similarly, the –CH2 bending vibration absorption peak shifted from 1460 cm−1 to 1427 cm−1. Peaks at bands 1427 and 1359 cm−1 are due to the bending of -CH2 and C-H, respectively, at C-6. The presence of peaks in CMF at bands 1336, 1359, 1201, 1165, 1106, 1059, 1034, and > 897 cm−1 are assigned to the in-plane bending of C–O–H (at C-2 and C-3), –CH2 wagging (at C-6), in-plane bending of C–O–H (at C-6), stretching of C–O–C (at β-glycoside bond), in-plane ring stretching, C–O stretching at C-3, C-O stretching (at C-6), and stretching of C–O–C (at β-glycoside bond) and C–CH (at C-5 and C-6), respectively. Bands at 710 cm−1 present in all 4 CMF samples were assigned to Iβ (monoclinic) phases cellulose (Sugiyama et al. 1991).
Figure 6 shows the Raman spectra and band detection from raw and treated samples (S1, S2, S3 and S4). The Raman bands observed in the cellulose sample were attributed primarily to the major cell wall components including cellulose, hemicelluloses and lignin, according to previous research on Raman spectra (Wiley and Atalla 1987). The band height indicated the concentration of cell wall components, i.e. the higher the band height, the greater the cell wall component concentrations. The band around 1500 cm−1 was attributed to the inward movement of the methylene group, whereas the band around 1430–1500 cm−1 was ascribed to the bending of HCH. The high intensities in the Raman spectra were obtained at band 1350–1430 cm−1 of COH bending and band 800–1180 cm−1 of CC and CO stretching with HCC and HCO bending. Band 150–550 cm−1 was assigned to CCC, COC, OCC and OCO stretching group with CC and CO motion present in the glucopyranose ring. The stretching band at region 1270–1350 cm−1 represented the bending of HCC and HCO (Szymańska-Chargot et al. 2011). It can be concluded that the FTIR and Raman spectrum of all treated samples were identical, and there was no significant differences observed amongst samples with shorter or longer bleaching times. Meanwhile, some characteristic peaks of raw PALF assigned to hemicellulose and lignin components were completely absent in the final bleached CMF. This finding also supported the typical spectrum of cellulose 1 (Lu and Hsieh 2012) (Fig. 7).

FTIR spectra of cellulose microfibres from pineapple leaf fibre (PALF) and treated cellulose fibres (S1, S2, S3 and S4)

Raman spectra of cellulose microfibres from treated cellulose fibres (S1, S2, S3 and S4)
Conclusions
This study was concerned with how the degree of individualisation affects the PALF morphology, chemical composition and structural arrangement of the CMF as the first step of an extensive study. The chemical treatments applied resulted in the individualised PALF microfibres, and bleaching treatment formed a network structure of CMF. With crystallite size of 3.564–3.807 nm, the prepared CMF need to be studied extensively for their efficient utilisation with nano-sized fibres. This study also showed alkali treatment and bleaching as viable methods to produce CMF. After the characterisation, the bleached cellulose after 4 h was cellulose native. Treatment with NaOH and bleaching were not able to convert cellulose I to cellulose II. Alpha cellulose from the treated method has a high crystallinity (85.41%) compared to the untreated sample. A monoclinic structure (I) was dominant in extensively long bleached samples as indicated by XRD diffractogram and FTIR-Raman spectra. Thus, all the findings obtained in the present study are very important for the efficient production of CMF for fabrication of new nanocomposites for various applications: from high-volume products such as cement, automotive parts, packaging, absorbent products (diapers), textile and precast concrete, to low-volume products including aerogels, paints, air and water filtration, cosmetics, drug delivery media, electronics, additive manufacturing, bone and tissue scaffolding, and thermoset adhesives.
References
Abraham E, Deepa B, Pothan L, Jacob M, Thomas S, Cvelbar U, Anandjiwala R (2011) Extraction of nanocellulose fibrils from lignocellulosic fibres: a novel approach. Carbohyd Polym 86:1468–1475
Acharjee TC, Jiang Z, Haynes RD, Lee YY (2017) Evaluation of chlorine dioxide as a supplementary pretreatment reagent for lignocellulosic biomass. Bioresour Technol 244:1049–1054
Akhtar N, Goyal D, Goyal A (2015) Biodegradation of cellulose and agricultural waste material. Advances in Biodegradation and Bioremediation of Industrial Waste. CRC Press, Boca Raton
Alves VD, Freitas F, Costa N, Carvalheira M, Oliveira R, Gonçalves MP, Reis MA (2010) Effect of temperature on the dynamic and steady-shear rheology of a new microbial extracellular polysaccharide produced from glycerol byproduct. Carbohyd Polym 79:981–988
Asim M, Abdan K, Jawaid M, Nasir M, Dashtizadeh Z, Ishak M, Hoque ME (2015) A review on pineapple leaves fibre and its composites. Int J Polym Sci 2015:1–16
Atalla RH, Vanderhart DL (1984) Native cellulose: a composite of two distinct crystalline forms. Science 223:283–285
Aziz FA et al (2016) The treated cellulose micro/nano fibers (CMNF) from bioresources in malaysia. Mater Sci Forum 846:434–439
Azraaie N et al (2016) Cellulose microfibrils/nanofibrils (CMNF) produced from banana (Musa acuminata) pseudo-stem wastes: isolation and characterization. Mater Sci Forum 846:448–453
Buffiere J, Balogh-Michels Z, Borrega M, Geiger T, Zimmermann T, Sixta H (2017) The chemical-free production of nanocelluloses from microcrystalline cellulose and their use as Pickering emulsion stabilizer. Carbohyd Polym 178:48–56
Cai J, Zhang L (2005) Rapid dissolution of cellulose in LiOH/urea and NaOH/urea aqueous solutions. Macromol Biosci 5:539–548
Chakraborty A, Sain M, Kortschot M (2005) Cellulose microfibrils: a novel method of preparation using high shear refining and cryocrushing. Holzforschung 59:102–107
Chandrasekhar S, Satyanarayana K, Pramada P, Raghavan P, Gupta T (2003) Review processing, properties and applications of reactive silica from rice husk—an overview. J Mater Sci 38:3159–3168
Chen W, Yu H, Liu Y, Chen P, Zhang M, Hai Y (2011) Individualization of cellulose nanofibers from wood using high-intensity ultrasonication combined with chemical pretreatments. Carbohyd Polym 83:1804–1811
Chen Y, Fan D, Han Y, Li G, Wang S (2017) Length-controlled cellulose nanofibrils produced using enzyme pretreatment and grinding. Cellulose 24:5431–5442
Cherian BM, Leão AL, de Souza SF, Thomas S, Pothan LA, Kottaisamy M (2010) Isolation of nanocellulose from pineapple leaf fibres by steam explosion. Carbohyd Polym 81:720–725
Cherian BM et al (2011) Cellulose nanocomposites with nanofibres isolated from pineapple leaf fibers for medical applications. Carbohyd Polym 86:1790–1798
Deepa B et al (2015) Utilization of various lignocellulosic biomass for the production of nanocellulose: a comparative study. Cellulose 22:1075–1090
dos Santos RM, Neto WPF, Silvério HA, Martins DF, Dantas NO, Pasquini D (2013) Cellulose nanocrystals from pineapple leaf, a new approach for the reuse of this agro-waste. Ind Crops Prod 50:707–714
Fan LT, Gharpuray MM, Lee Y-H (1987) Nature of cellulosic material. Cellulose Hydrolysis. Springer, Berlin
Fawcett TG, Faber J, Crane D, Kabekkodu S, Sagnella D, Blanton J (2008) A reference diffraction database for non-crystalline, partially crystalline and amorphous materials. International Centre for Diffraction Data (ICDD) Polymer Subcommittee Meeting
French AD (2014) Idealized powder diffraction patterns for cellulose polymorphs. Cellulose 21:885–896
French AD, Santiago Cintrón M (2013) Cellulose polymorphy, crystallite size, and the Segal crystallinity index. Cellulose 20:583–588
Ibrahim NA, Azraaie N, Abidin Z, Mohd NA, Razali NAM, Aziz FA, Zakaria S (2014) Preparation and characterization of alpha cellulose of pineapple (Ananas comosus) leaf fibres (PALF). Adv Mat Res 2014:147–150
Ishak WHW, Yin OS, Ahmad I (2016) Effect of micro-and nano-size of cellulose particles extracted from rice husk on the properties of hydrogels. Polym Polym Compos 24:783
Jin X-J, Pascal Kamdem D (2009) Chemical composition, crystallinity and crystallite cellulose size in populus hybrids and aspen. Cellulose Chem Technol 43:229
Khalil HA, Bhat A, Yusra AI (2012) Green composites from sustainable cellulose nanofibrils: a review. Carbohyd Polym 87:963–979
Khelifa F, Habibi Y, Dubois P (2016) Nanocellulose-based polymeric blends for coating applications. Multifunctional Polymeric Nanocomposites Based on Cellulosic Reinforcements. William Andrew: Applied Science, Oxford
Klemm D, Kramer F, Moritz S, Lindström T, Ankerfors M, Gray D, Dorris A (2011) Nanocelluloses: a new family of nature-based materials. Angew Chem Int Ed 50:5438–5466
Kondo T (1997) The assignment of IR absorption bands due to free hydroxyl groups in cellulose. Cellulose 4:281
Li S, Lee PS (2017) Development and applications of transparent conductive nanocellulose paper. Sci Technol Adv Mater 18:620–633
Li J et al (2012) Homogeneous isolation of nanocellulose from sugarcane bagasse by high pressure homogenization. Carbohyd Polym 90:1609–1613
Lizundia E et al (2016) PLLA-grafted cellulose nanocrystals: role of the CNC content and grafting on the PLA bionanocomposite film properties. Carbohyd Polym 142:105–113
Łojewska J, Miśkowiec P, Łojewski T, Proniewicz L (2005) Cellulose oxidative and hydrolytic degradation: in situ FTIR approach. Polym Degrad Stab 88:512–520
Lu P, Hsieh Y-L (2012) Preparation and characterization of cellulose nanocrystals from rice straw. Carbohyd Polym 87:564–573
Ludwicka K, Jedrzejczak-Krzepkowska M, Kubiak K, Kolodziejczyk M, Pankiewicz T, Bielecki S (2017) Medical and cosmetic applications of bacterial nanocellulose. Bacterial Nanocellulose. Elsevier, Netherlands
Messiry M (2014) Morphological analysis of micro-fibrillated cellulose from different raw materials for fiber plastic composites. J Text Sci Eng 4:1
Mohamed A, Sapuan S, Shahjahan M, Khalina A (2010) Effects of simple abrasive combing and pretreatments on the properties of pineapple leaf fibers (palf) and palf-vinyl ester composite adhesion. Polym Plast Technol 49:972–978
Mohammadkazemi F, Doosthoseini K, Ganjian E, Azin M (2015) Manufacturing of bacterial nano-cellulose reinforced fiber − cement composites. Constr Build Mater 101:958–964
Moon RJ, Martini A, Nairn J, Simonsen J, Youngblood J (2011) Cellulose nanomaterials review: structure, properties and nanocomposites. Chem Soc Rev 40:3941–3994
Mtibe A, Linganiso LZ, Mathew AP, Oksman K, John MJ, Anandjiwala RD (2015) A comparative study on properties of micro and nanopapers produced from cellulose and cellulose nanofibres. Carbohyd Polym 118:1–8
Nam S, French AD, Condon BD, Concha M (2016) Segal crystallinity index revisited by the simulation of X-ray diffraction patterns of cotton cellulose Iβ and cellulose II. Carbohyd Polym 135:1–9
Neto ARS et al (2015) Comparative study of 12 pineapple leaf fiber varieties for use as mechanical reinforcement in polymer composites. Ind Crops Prod 64:68–78
Nishiyama Y, Langan P, Chanzy H (2002) Crystal structure and hydrogen-bonding system in cellulose Iβ from synchrotron X-ray and neutron fiber diffraction. J Am Chem Soc 124:9074–9082
Oh SY, Yoo DI, Shin Y, Seo G (2005) FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohyd Res 340:417–428
Owolabi AF, Haafiz MM, Hossain MS, Hussin MH, Fazita MN (2017) Influence of alkaline hydrogen peroxide pre-hydrolysis on the isolation of microcrystalline cellulose from oil palm fronds. Int J Biol Macromol 95:1228–1234
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3:10
Rambabu N, Panthapulakkal S, Sain M, Dalai A (2016) Production of nanocellulose fibers from pinecone biomass: evaluation and optimization of chemical and mechanical treatment conditions on mechanical properties of nanocellulose films. Ind Crops Prod 83:746–754
Rosa M et al (2010) Cellulose nanowhiskers from coconut husk fibers: effect of preparation conditions on their thermal and morphological behavior. Carbohyd Polym 81:83–92
Sabo R, Yermakov A, Law CT, Elhajjar R (2016) Nanocellulose-enabled electronics, energy harvesting devices, smart materials and sensors: a review. J Renew Mater 4:297–312
Sanoj P, Balasubramanian K (2014) High performance structural nano cellulose composites for motor vehicle spring suspension system. Int J Plast Technol 18:383–389
Serpa A, Velásquez-Cock J, Gañán P, Castro C, Vélez L, Zuluaga R (2016) Vegetable nanocellulose in food science: a review. Food Hydrocoll 57:178–186
Shamsudin R, Daud WRW, Takriff MS, Hassan O (2007) Physicochemical properties of the Josapine variety of pineapple fruit. Int J Food Eng 3:1–12
Sharma S, Nair SS, Zhang Z, Ragauskas AJ, Deng Y (2015) Characterization of micro fibrillation process of cellulose and mercerized cellulose pulp. RSC Adv 5:63111–63122
Shi Z, Zhang Y, Phillips GO, Yang G (2014) Utilization of bacterial cellulose in food. Food Hydrocoll 35:539–545
Stephen B, Parungao MM (2003) Production of wood-decay enzymes, mass loss and lignin solubilization in wood by tropical Xylariaceae. Mycol Res 107:231–235
Sugiyama J, Persson J, Chanzy H (1991) Combined infrared and electron diffraction study of the polymorphism of native celluloses. Macromolecules 24:2461–2466
Szymańska-Chargot M, Cybulska J, Zdunek A (2011) Sensing the structural differences in cellulose from apple and bacterial cell wall materials by Raman and FT-IR spectroscopy. Sensors 11:5543–5560
Tang J, Li X, Bao L, Chen L, Hong FF (2017) Comparison of two types of bioreactors for synthesis of bacterial nanocellulose tubes as potential medical prostheses including artificial blood vessels. J Chem Technol Biotechnol 92:1218–1228
Tappi T (2002) Technical Association of the Pulp and Paper Industry, TAPPI 222 om-06: Acid-insoluble lignin in wood and pulp. Angewandte Chemie International Edition
Wada M, Okano T, Sugiyama J (1997) Synchrotron-radiated X-ray and neutron diffraction study of native cellulose. Cellulose 4:221–232
Wang S, Cheng Q (2009) A novel process to isolate fibrils from cellulose fibers by high-intensity ultrasonication, Part 1: process optimization. J Appl Polym Sci 113:1270–1275
Wang W, Sabo RC, Mozuch MD, Kersten P, Zhu J, Jin Y (2015) Physical and mechanical properties of cellulose nanofibril films from bleached eucalyptus pulp by endoglucanase treatment and microfluidization. J Polym Environ 23:551–558
Werner A, Schmitt V, Sèbe G, Héroguez V (2017) Synthesis of surfactant-free micro-and nanolatexes from Pickering emulsions stabilized by acetylated cellulose nanocrystals. Polym Chem 8:6064–6072
Wiley JH, Atalla RH (1987) Band assignments in the Raman spectra of celluloses. Carbohyd Res 160:113–129
Wise LE, Murphy M, Adieco AAD (1946) A chlorite holocellulose, its fractionation and bearing on summative wood analysis and studies on the hemicelluloses. Paper Trad J 122:35–43
Ya’acob WMHW, Aziz FA (2017) Nanocellulose: a promising material for engineering-an overview. Int J Mater Eng Innov 8:71–86
Yue Y, Han G, Wu Q (2013) Transitional properties of cotton fibers from cellulose I to cellulose II structure. BioResources 8:6460–6471
Zugenmaier P (2001) Conformation and packing of various crystalline cellulose fibers. Prog Polym Sci 26:1341–1417
Acknowledgment
This study was carried out in collaboration with the Forest Research Institute of Malaysia (FRIM), Universiti Pertahanan Nasional Malaysia (UPNM) and members of the UPNM X-ray Research Group. The authors would like to acknowledge the financial support provided by The Ministry of Higher Education under the Niche Research Grant Scheme (NRGS) NRGS/2013/UPNM/PK/P1.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fareez, I.M., Ibrahim, N.A., Wan Yaacob, W.M.H. et al. Characteristics of cellulose extracted from Josapine pineapple leaf fibre after alkali treatment followed by extensive bleaching. Cellulose 25, 4407–4421 (2018). https://doi.org/10.1007/s10570-018-1878-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-1878-0